|
|

Estructura genética del “roble belloto” Quercus skinneri (Fagaceae) en El Salvador
Roberto Antonio Navarro-Linares1, 2![]() ,
Ligia Elena Muñoz-Molina1, 2
,
Ligia Elena Muñoz-Molina1, 2 ![]() &
Miguel Ángel Moreno-Mendoza1, 2
&
Miguel Ángel Moreno-Mendoza1, 2 ![]()
1. Universidad de El Salvador, Escuela de Biología, Facultad de Ciencias Naturales y Matemática, Ciudad Universitaria "Dr. Fabio Castillo Figueroa", Final de Av. Mártires y Héroes del 30 julio, San Salvador, El Salvador; roberto.navarro@ues.edu.sv; ligia.molina@ues.edu.sv; miguel.moreno@ues.edu.sv
2. Universidad de El Salvador, Grupo de Investigación en Bioinformática Estructural, Biomodelos y Biomarcadores, Facultad de Ciencias Naturales y Matemática, Ciudad Universitaria "Dr. Fabio Castillo Figueroa", Final de Av. Mártires y Héroes del 30 julio, San Salvador, El Salvador.
Recibido 21-IV-2025 · Corregido 09-VII-2025 · Aceptado 22-VII-2025
DOI: https://doi.org/10.22458/urj.v17i1.5762
ABSTRACT. “Genetic Structure of the ‘roble belloto’ Quercus skinneri (Fagaceae) in El Salvador”. Introduction: In El Salvador, Quercus skinneri Benth. is restricted to high-altitude temperate areas. The lack of genetic studies limits conservation decision-making, despite the importance of genetic diversity in these forests. Objective: To determine the genetic variability and structure of Q. skinneri populations in three localities of El Salvador. Methods: Samples were collected between July and December 2020. We assessed the genetic variability of the tree by collecting leaves from ten individuals per locality, and sequencing and analyzing a total of 19 individuals using two DNA barcode regions: a nuclear region (ITS2) and a plastid region (trnH-psbA). Based on aligned sequences, we calculated genetic diversity indices, population structure, isolation by distance, and phylogeny. Results: Genetic diversity was higher in ITS2 (π = 0.01576; Hd = 0,90643; h = 10) than in trnH-psbA (π = 0,00519; Hd = 0,48538; h = 3). Both regions showed populational structure, with the San Vicente Volcano population clearly differentiated (FST = 0,79972–1) from the others, as reflected in haplotype maps and phylogenetic trees. Conclusion: ITS2 and trnH-psbA differed in their ability to detect genetic variability in Q. skinneri. Both revealed populational structure, notably the differentiation of San Vicente Volcano, suggesting distinct genetic lineages.
Keywords: genetic diversity, phylogeny, oak forests, genetic differentiation.
RESUMEN. Introducción: En El Salvador, Quercus skinneri Benth. está restringido a zonas templadas de altitud. La falta de estudios genéticos limita la toma de decisiones para su conservación, a pesar de la importancia de la diversidad genética en estos bosques. Objetivo: Determinar la variabilidad y estructura genética de poblaciones de Q. skinneri en tres localidades de El Salvador. Métodos: Muestreamos entre julio y diciembre de 2020. Evaluamos la variabilidad genética del árbol tomando hojas de diez individuos por localidad; secuenciamos y analizamos un total de 19 individuos, utilizando dos regiones de códigos de barras de ADN: una región nuclear (ITS2) y una región plastídica (trnH-psbA). A partir de las secuencias alineadas, calculamos índices de diversidad genética, estructura poblacional, aislamiento por distancia y filogenia. Resultados: La diversidad genética fue mayor en ITS2 (π = 0,01576; Hd = 0,90643; h = 10) que en trnH-psbA (π = 0,00519; Hd = 0,48538; h = 3). Ambas regiones tienen una estructura poblacional, con la población del Volcán de San Vicente claramente diferenciada (FST = 0,79972–1) de las demás, lo cual se reflejó en los mapas de haplotipos y en los árboles filogenéticos. Conclusión: ITS2 y trnH-psbA difirieron en su capacidad para detectar variabilidad genética en Q. skinneri. Ambas revelaron estructura poblacional, destacando la diferenciación del Volcán de San Vicente, lo que sugiere linajes genéticos distintos.
Palabras clave: diversidad genética, filogenia, bosque de robles, diferenciación genética.
En las últimas décadas, los estudios en genética de poblaciones han adquirido una importancia prioritaria debido a su papel fundamental para la conservación de la biodiversidad, especialmente en el contexto de las especies vegetales. Estudios demuestran la importancia que tiene la diversidad genética en las posibilidades de adaptación y supervivencia en especies vegetales, estando relacionada a factores como la resistencia a enfermedades y plagas, o a la capacidad de sobrellevar cambios en los regímenes climáticos de su hábitat (Jump et al., 2009). A su vez, la información obtenida de los estudios genéticos son una herramienta que permiten el diseño e implementación efectiva de unidades de conservación prioritarias, programas de restauración y reintroducción que aseguren que las poblaciones restablecidas posean la variabilidad genética necesaria para afrontar las amenazas a las que están expuestas (Hvilsom et al., 2022).
El género Quercus L. está compuesto por árboles y arbustos que se distribuyen por gran parte del hemisferio norte, llegando a latitudes tropicales tanto en América como en el sudeste asiático, son uno de los grupos de árboles más diversos y significativos ecológicamente a nivel mundial, con más de 500 especies reportadas, siendo México un importante centro de diversidad, con más de 150 especies reportadas, en su mayoría endémicas (Morales-Saldaña et al., 2022; Valencia-A., 2017). Los robles juegan un papel fundamental en sus ecosistemas, prestando diversos servicios ecológicos, entre los que se pueden mencionar captación de agua lluvia y atmosférica, formación del suelo, captación de dióxido de carbono y reducción de la erosión, así como proveer de una gran cantidad de alimento para diferentes especies por medio del fenómeno de su fructificación masiva conocido como masting (Rodríguez-Acosta & Coombes, 2020). En El Salvador, el género Quercus L. se encuentra distribuido en las principales elevaciones del país, desde los 900 hasta los 2 300 m.s.n.m., siendo su presencia importante en los bosques de pino-encino, pino-encino-liquidámbar, robledales y bosques nebulosos. Actualmente se registran 16 especies de Quercus L. en el país, distribuyéndose en 12 de los 14 departamentos, de estas especies, Quercus skinneri Benth. sobresale por presentar la distribución más amplia, con registros en cinco departamentos (Berendsohn et al., 2009; Lauer, 1954). Esta especie se caracteriza por poseer hojas delgadas y membranosas, con margen que presenta de diez a 13 dientes largo-aristados, con envés glabro o con escasos tricomas, bellotas ovoides, de 15 a 50mm de largo por 20 a 50mm de diámetro, de color castaño, incluidas un cuarto o menos del largo en las cúpulas (Romero Rangel, 2006). En cuanto a su estado de conservación, la IUCN lo clasifica como una especie casi amenazada (NT), ya que, a pesar de su amplia distribución en México y Centroamérica, las elevadas tasas de deforestación en las últimas décadas han llevado a la reducción y fragmentación de sus poblaciones (Jerome, 2020). Si bien, no se cuentan con datos certeros sobre el tamaño de sus poblaciones en El Salvador, algunas de las amenazas que contribuyen a la reducción en la abundancia del género Quercus L. son: la deforestación, cambios de uso de suelo para agricultura, modificaciones del sotobosque para el cultivo de café y cacao, así también el aumento del turismo descontrolado en áreas de clima frio en El Salvador(Good et al., 2024).
La técnica de los códigos de barra de ADN (DNA barcodes) propuso originalmente el uso de una secuencia corta y estandarizada de ADN mitocondrial, el gen COI, como una herramienta para identificar y discriminar especies animales (Hebert et al., 2003). Posteriormente, los avances en el establecimiento de un código de barras para plantas llevaron a sugerir el uso de regiones del cloroplasto (matK, rbcL y trnH-psbA) y de la región nuclear ITS, adoptándose la combinación matK + rbcL como barcode universal (Hollingsworth, 2011; Hollingsworth et al., 2009; Kress et al., 2005). En años recientes, nuevas regiones utilizadas como barcodes han demostrado una gran utilidad. Un ejemplo de ello es la región ITS2, utilizada en estudios de identificación molecular, medición de la diversidad genética y determinación de filogenias en diversas familias, como Rosaceae (Pang et al., 2011), Selaginellaceae (Gu et al., 2013), Orchidaceae (Feng et al., 2015), Solanaceae (Feng et al., 2016) Musaceae (Dhivya et al., 2020) y Pinaceae (Sokołowska et al., 2022), así como en espermatofitas en general (Qin et al., 2017). De manera complementaria, la región del cloroplasto trnH-psbA ha evidenciado una notable eficacia como código de barras, siendo empleada en estudios de identificación molecular, control comercial y análisis de diversidad genética en diversas familias, incluyendo a Fabaceae y Myrtaceae en especies arbóreas amenazadas del Bosque Atlántico (Bolson et al., 2015), Poaceae y Fabaceae en la identificación de especies forrajeras (Loera-Sánchez et al., 2020), así como en la autenticación de plantas melíferas en ecosistemas tropicales de Yucatán (Durán Escalante et al., 2023).
Si bien la secuenciación de próxima generación (NGS) ha revolucionado el estudio del género Quercus L. al revelar procesos evolutivos complejos, como, patrones globales de diversificación y flujo génico (Hipp et al., 2020), especiación simpátrica en clados americanos (Hipp et al., 2018), y adaptación local mediada por variantes climáticas (Martins et al., 2018), los códigos de barras de ADN representan una alternativa económica y versátil para abordar preguntas clave en el género. Como ejemplos tenemos el uso de marcadores de trnH-psbA y matK, que permitió reconstruir historias filogeográficas en robles japoneses (Q. mongolica var. crispula), exponiendo estructuras poblacionales moldeadas por cambios climáticos del Cuaternario (Okaura et al., 2007), asimismo, la región ITS2 y sus regiones circundantes demostraron alta eficacia para discriminar especies de Quercus L. en Turquía, destacando su utilidad en identificación taxonómica (Aykut, 2020), mientras que la combinación de rbcL + matK + ITS2 resolvió relaciones filogenéticas en robles mexicanos con resolución robusta (Pacheco-Reyes et al., 2021), demostrando así la utilidad de este tipo de marcadores en el estudio de robles.
MATERIALES Y MÉTODOS
Toma de muestras: Recolectamos las muestras entre julio y diciembre del año 2020, con permisos del Ministerio de Medio Ambiente y Recursos Naturales de El Salvador, Resolución -MARN-DEB-GVS-AIMA-013-2020, en tres sitios de estudio: Laguna de las Ninfas, Cerro Verde y Volcán de San Vicente (Fig. 1). Realizamos un muestreo dirigido, recolectando cinco hojas de los primeros diez árboles encontrados, asegurando una distancia mínima de diez metros entre cada individuo, para un total de 30 individuos. Cada hoja fue limpiada con etanol al 70% para eliminar contaminantes ambientales, posteriormente se colocaron en bolsas de papel y se introdujeron en un desecador con gel de sílice, con el fin de deshidratar el material vegetal rápidamente (Fernández et al., 2000). Herborizamos al menos dos muestras fértiles por sitio, así como una muestra estéril por individuo, las cuales fueron depositadas en la colección del herbario del Instituto de Investigaciones Tropicales de la Universidad de El Salvador, con sus respectivos metadatos.
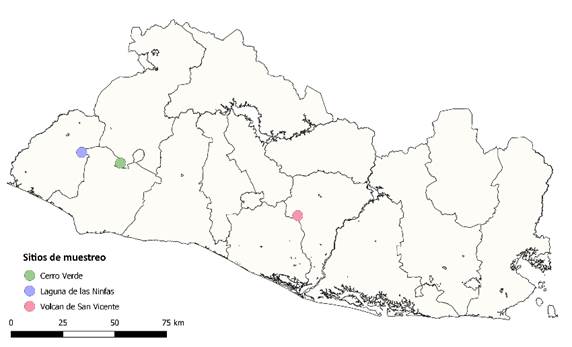
Fig. 1: Ubicación de los sitios de recolecta de muestras de Quercus skinneri.
Extracción de ADN y verificación de calidad: Utilizamos una modificación del método CTAB con los siguientes ajustes en el buffer de extracción: 10mM Tris-HCl (pH 8.0), 20mM EDTA (pH 8.0), 1.8M NaCl, 2% CTAB, 1% PVP-40 y 1% β-mercaptoetanol. Realizamos dos lavados con cloroformo-alcohol isoamílico (24:1; v/v), siguiendo el protocolo descrito por Fernández (2000). Verificamos la calidad del ADN extraído mediante electroforesis, utilizando geles de agarosa al 1%, a su vez, se midió la concentración del ADN por absorbancia, utilizando el espectrofotómetro Jenway 737501.
Amplificación por PCR: Amplificamos dos regiones, la región ITS2, que corresponde al segundo espaciador transcrito interno del ribosoma del genoma nuclear y la región no codificante del cloroplasto trnH-psbA. Para la amplificación de la región ITS2 utilizamos los cebadores F: ATGCGATACTTGGTGTGAAT (Chen et al., 2010) y R: TCCTCCGCTTATTGATATGC (White et al., 1990), a su vez, utilizamos los cebadores F: ACTGCCTTGATCCACTTGGC y R: CGAAGCTCCATCTACAAATGG para la región trnH-psbA (Hamilton, 1999). Llevamos a cabo la PCR en un termociclador MultiGene Mini TC020-24, utilizando la master mix GoTaq® G2 (Promega, USA) con los siguientes componentes: 17,5µL de master mix, 0,7µL de cada cebador (0,2µM) y 20ng de ADN total, agregamos agua ultrapura hasta completar un volumen 35µL por reacción. La amplificación inició con un ciclo de desnaturalización de 5min a 94°C, seguido de 35 ciclos de 45s de desnaturalización a 95°C, 45s de alineamiento a 59°C (ITS2) o 61°C (trnH-psbA), 40s de extensión a 74°C y un ciclo de extensión final a 74°C por 7min. El éxito de la amplificación se corroboró mediante electroforesis, utilizando un gel de agarosa al 2%.
Secuenciación y procesamiento: La secuenciación se llevó a cabo por medio de la empresa MACROGEN Inc. (Corea) donde se realizó la purificación de los productos de PCR previo a la secuenciación Sanger, la cual se efectuó en el secuenciador de ADN ABI 3730XL (Applied Biosystems®). Utilizamos el software Geneious Prime 2024.07 para el procesamiento de las secuencias crudas, en este eliminamos los extremos inespecíficos de las secuencias y se descartaron aquellas secuencias que presentaron una mala calidad (puntuación menor a 40 en el gráfico Phred) en la mayor parte de sus bases (Ewing & Green, 1998), utilizamos el mismo software para obtener las secuencias consenso de cada muestra. Obtuvimos 25 secuencias consenso de buena calidad con un tamaño de 288pb para la región ITS2 y 24 secuencias consenso de un tamaño de 398pb de buena calidad para la región trnH-psbA. Realizamos un alineamiento de secuencias múltiples utilizando el algoritmo MUSCLE, incluido en el software MEGA-12 12.0.7. (Kumar et al., 2024). Una vez alineadas las secuencias consenso, se decidió analizar solamente la información de los individuos que tuvieran secuencias para ambos marcadores, siendo estos solamente 19 individuos. Las secuencias analizadas para cada región pueden consultarse en: Material Suplementario 1.
Análisis de diversidad genética y estructuración poblacional: Calculamos la diversidad nucleotídica (π), el número de sitios segregantes (S), la frecuencia de haplotipos (H), la diversidad haplotípica (Hd) y la varianza de k utilizando el software DnaSP v6.12.03 (Rozas et al., 2017). Evaluamos la estructuración poblacional mediante la comparación de la varianza estocástica de k, la varianza de muestreo y un Análisis de Varianza Molecular (AMOVA), seguido del cálculo de los índices de fijación FST utilizando el software Arlequin v3.5.2.2 (Excoffier et al., 2017).
Como representación gráfica de las diferencias entre haplotipos, se generó un mapa de haplotipos mediante el método Median Joining Network en el programa PopArt (Leigh & Bryant, 2015).
Análisis de aislamiento por distancia: Comprobamos la hipótesis de aislamiento por distancia por medio de la prueba de Mantel, utilizando la extensión GenAlEx 6.503 para MS Excel 2016 (Peakall & Smouse, 2012). Para ello, se comparó la matriz de distancias FST con la matriz de distancias geográficas, generada con el software Geographic Distance Matrix Generator v1.2.3 (Ersts, 2025).
Análisis filogenético: Para el análisis filogenético, tomamos los sets de 19 secuencias para cada región y se agregó una secuencia outgroup, la cual obtuvimos mediante el uso de BLAST, escogiendo a la especie Castanea dentata (Marshall) Borkh., de la cual se encontraron secuencias de ambas regiones, utilizando las accesiones MK093982.1 y KP643301.1 para las regiones ITS2 y trnH-psbA, respectivamente. Utilizamos el método de máxima verosimilitud para la realización de nuestro árbol filogenético. Primeramente, calculamos las puntuaciones del criterio de información bayesiana (BIC) y del criterio de información de Akaike (AIC) para elegir el modelo de sustitución más adecuado para cada set de secuencias, siendo el modelo Tamura-3 + I el más adecuado para ambos sets de secuencias. Una vez seleccionado el modelo, seleccionamos 1 000 replicaciones bootstrap para mejorar la confiabilidad del árbol, todos estos procesos se realizaron utilizando el software MEGA-12 (Kumar et al., 2024), finalmente para la mejor presentación de los datos filogenéticos se utilizó la aplicación tvBOT (Xie et al., 2023)
RESULTADOS
Diversidad genética: La región ITS2 presentó una mayor diversidad nucleotídica, un mayor número de sitios segregantes, así como un mayor número de haplotipos y mayor diversidad haplotípica en comparación con la región trnH-psbA. En ITS2, los sitios Laguna de las Ninfas y Cerro Verde mostraron índices de diversidad genética similares entre sí, ambos superiores a los observados en el sitio Volcán de San Vicente. En contraste, para la región trnH-psbA, únicamente la población de Laguna de las Ninfas presentó más de un haplotipo, siendo esta la única localidad que mostró valores mayores a cero para el número de sitios segregantes, diversidad nucleotídica y diversidad haplotípica (Tabla 1). La varianza estocástica obtenida para ITS2 fue Vst(k) = 1,737, mientras que la varianza de muestreo fue menor, con un valor de Vs(k) = 0,183. Para la región trnH-psbA, la varianza estocástica fue Vst(k) = 0,563, y la varianza de muestreo fue también menor, Vs(k) = 0,059.
TABLA 1
Índices de diversidad nucleotídica en las regiones ITS2 y trnH-psbA de Quercus skinneri.
|
Sitio |
# individuos |
ITS2 S |
π |
H |
Hd |
trnH-psbA S |
π |
H |
Hd |
|
Cerro Verde |
7 |
6 |
0,00794 |
5 |
0,85714 |
0 |
0 |
1 |
0 |
|
Laguna de las Ninfas |
7 |
6 |
0,00794 |
5 |
0,85714 |
4 |
0,00288 |
2 |
0,28571 |
|
Volcán de San Vicente |
5 |
2 |
0,00278 |
3 |
0,70000 |
0 |
0 |
1 |
0 |
|
Total |
19 |
13 |
0,01576 |
10 |
0, 90643 |
8 |
0,00519 |
3 |
0,48538 |
S=número de sitios segregantes, π = diversidad nucleotídica, H = número de haplotipos, Hd = diversidad haplotípica.
Análisis de Varianza Molecular (AMOVA): El análisis de varianza molecular indica la existencia de estructuración poblacional para los marcadores ITS2 y trnH-psbA, ya que, el porcentaje de variación entre las poblaciones, 69,22% y 85,90%, es mayor que el porcentaje de variación dentro de las poblaciones, 30,77% y 14,14%, respectivamente.
TABLA 2
Valores del Análisis de Varianza Molecular para la región ITS2 y trnH-psbA
|
ITS2 |
||||
|
Suma de cuadrados |
Componentes de variación |
Porcentaje de variación |
P-valor |
|
|
Entre poblaciones |
51,056 |
1,96807 |
57,49 |
0,00000 |
|
Dentro de las poblaciones |
30,629 |
0,87510 |
42,51 |
0,00000 |
|
Total |
81,684 |
2,84318 |
||
|
trnH-psbA |
||||
|
Suma de cuadrados |
Componentes de variación |
Porcentaje de variación |
P-valor |
|
|
Entre poblaciones |
30,195 |
1,18964 |
85,90 |
0,00000 |
|
Dentro de las poblaciones |
6,857 |
0,19592 |
14,14 |
0,00000 |
|
Total |
37,053 |
1,38556 |
||
Índice de fijación por pares FST: Tomando en cuenta las dos regiones analizadas, observamos con mayor claridad la estructuración poblacional basada en el índice de fijación FST. La población de Volcán de San Vicente es la única que presenta valores de diferenciación genética significativos en comparación con las otras dos poblaciones (Tabla 3; Tabla 4). Para la región ITS2, los valores de FST que obtuvimos fueron: Volcán de San Vicente: Cerro Verde = 0,79972, Volcán de San Vicente: Laguna de las Ninfas = 0,81984 y Cerro Verde: Laguna de las Ninfas = 0,07692, este último sin significancia estadística. En el caso del marcador trnH-psbA, observamos el mismo patrón, aunque las diferencias entre la población de Volcán de San Vicente y las demás poblaciones son mucho más marcadas, alcanzando valores máximos de diferenciación genética: Volcán de San Vicente: Cerro Verde = 1,000, Volcán de San Vicente:Laguna de las Ninfas = 0,86599. Por su parte, las poblaciones de Cerro Verde: Laguna de las Ninfas alcanzan un valor de FST igual al observado con la región ITS2 de 0,07692, el cual tampoco es significativo.
TABLA 3
Comparación por pares de índice de diferenciación genética FST para la región ITS2
|
Cerro Verde |
Volcán de San Vicente |
Laguna de las Ninfas |
|
|
Cerro Verde |
0,00000 |
— |
— |
|
Volcán de San Vicente |
0,79972* |
0,00000 |
— |
|
Laguna de las Ninfas |
0,07692 |
0,81984* |
0,00000 |
Valores significativos de diferenciación *cuando p<0.05.
TABLA 4
Comparación por pares de índice de diferenciación genética FST para la región trnH-psbA
|
Cerro Verde |
Volcán de San Vicente |
Laguna de las Ninfas |
|
|
Cerro Verde |
0,00000 |
— |
— |
|
Volcán de San Vicente |
1,00000* |
0,00000 |
— |
|
Laguna de las Ninfas |
0,07692 |
0,86599* |
0,00000 |
Valores significativos de diferenciación *cuando p<0.05.
Aislamiento por distancia: La prueba de Mantel mostró un coeficiente de correlación alto (Rxy = 0,982), aunque no estadísticamente significativo (p = 0,163). Esta baja significancia estadística esta probablemente relacionada con el bajo número de poblaciones comparadas (n = 3). La regresión lineal entre las variables presentó una relación exacta (R2 = 1).
Mapa de haplotipos: En el mapa de haplotipos se visualiza gráficamente la estructuración poblacional. Para la región ITS2 (Fig. 2A), se observa que los individuos de los sitios Cerro Verde y Laguna de las Ninfas comparten haplotipos, formando una red de conectividad entre ellos. Cada haplotipo se encuentra separado por un único paso mutacional, lo que indica una alta similitud genética. Además, tanto Cerro Verde como Laguna de las Ninfas presentan dos haplotipos privados, cada uno de ellos separado por dos pasos mutacionales del resto de los haplotipos. Al lado opuesto del árbol observamos los haplotipos pertenecientes a la población de Volcán de San Vicente los cuales no se relacionan directamente con el resto de los haplotipos.
El mapa de haplotipos para la región trnH-psbA (Fig. 2B) muestra una estructuración poblacional similar a la observada en la región ITS2. Los individuos de los sitios Cerro Verde y Laguna de las Ninfas comparten un solo haplotipo, exceptuando un único individuo perteneciente a Laguna de Las Ninfas, el cual forma su propio haplotipo y presenta una diferenciación similar a la del outgroup, de igual modo, los individuos del sitio Volcán de San Vicente comparten un único haplotipo privado, donde se agrupan todos sus individuos, lo que indica una diferenciación genética marcada con respecto a las demás poblaciones. Este mapa también evidencia que la región trnH-psbA es altamente conservada, presentando menor variación genética en comparación con ITS2. Los alineamientos de los haplotipos pueden consultarse en el ANEXO 1.
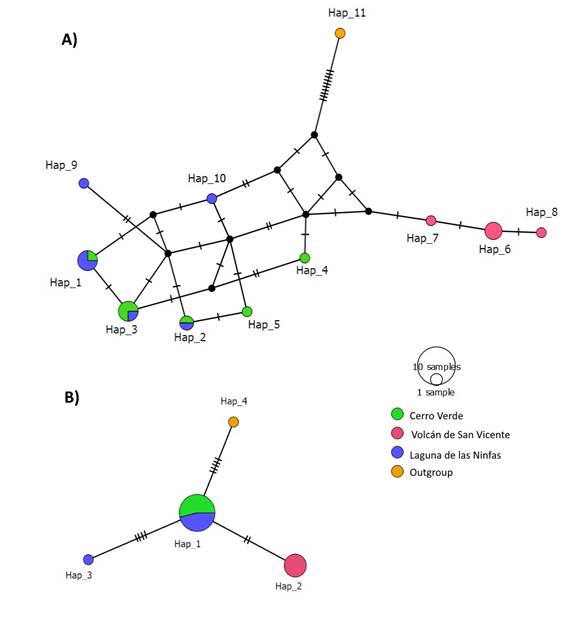
Fig. 2. Haplotipos de la región ITS2.
Análisis filogenético: El árbol filogenético de la región ITS2 nos muestra un agrupamiento de la mayoría de los individuos del occidente del país en un grupo, y los individuos del sitio Volcán de San Vicente en un grupo diferente, esto sigue el patrón observado en el mapa de haplotipos, sugiriendo que los individuos de los sitios Cerro Verde y Laguna de las Ninfas comparten un ancestro común más próximo entre ellos que con el sitio Volcán de San Vicente, de igual forma, el largo y ramificación de la parte superior del árbol concuerda con la mayor diversidad genética encontrada en las poblaciones occidentales (Fig. 3). De igual manera, el árbol efectuado sobre las secuencias de la región trnH-psbA nos muestra resultados similares a los encontrados en por los otros métodos, mostrando una poca diferenciación entre los individuos de las localidades Cerro Verde y Laguna de las Ninfas, formando en su mayoría un único grupo, y estas a su vez, estando diferenciadas de los individuos de la localidad Volcán de San Vicente. Cabe resaltar que un único individuo (LN1) se diferencia de las demás muestras de su localidad, teniendo un grado de diferenciación similar al del outgroup (Fig. 4).
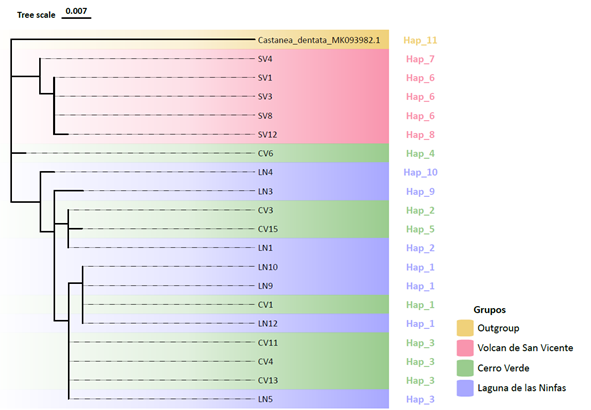
Fig. 3. Filogenia de la región ITS2.
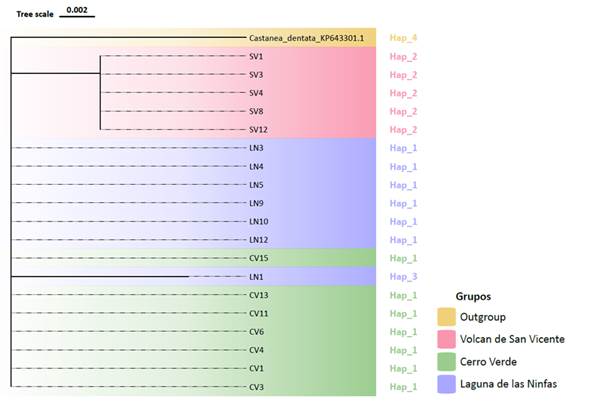
Fig. 4. Filogenia de la región trnH-psbA.
DISCUSIÓN
Al comparar ambas regiones, se observa una disparidad en los valores de diversidad genética. Para ITS2, el valor total fue π = 0,01576, mientras que para trnH-psbA fue considerablemente menor, con π = 0,00519. Los valores obtenidos para ITS2 son comparables a los reportados en otros estudios, como en Quercus ilex L. y Q. coccifera L. (Simeone et al., 2013). En contraste, los valores nulos o extremadamente bajos de diversidad genética en trnH-psbA son similares a los encontrados en Q. phillyreoides A.Gray en Japón, con valores de π = 0,00004 (Liu et al., 2013).
Se ha observado que la presencia de un mayor número de especies simpátricas de robles rojos (sección Lobatae) se asocia con una mayor diversidad genética en Quercus castanea Neé, tanto en regiones nucleares como cloroplásticas, lo que sugiere que procesos de hibridación y retrogresión podrían estar involucrados (Valencia-Cuevas et al., 2014). Esto concuerda con la mayor diversidad genética encontrada en los sitios Cerro Verde y Laguna de las Ninfas, que cohabita con tres especies de la sección Lobatae (Q. acatenangensis Trel., Q. benthamii A.DC. y Q. tristis Liebm.), en contraste, la única otra especie de roble reportada para el sitio Volcán de San Vicente es Q. vicentensis Trel., perteneciente a la sección Quercus, por lo que se espera que los individuos de este sitio tengan menor acceso a esta fuente de variación genética (Berendsohn et al., 2009).
Nuestros resultados demuestran un patrón de diferenciación entre los sitios de estudio analizados, mostrando diferenciación genética entre los individuos de Q. skinneri Benth. presentes la localidad Volcán de San Vicente con respecto a los encontrados en las localidades Cerro Verde y Laguna de las Ninfas, los cuales forman una única población desde el punto de vista genético, evidenciado por los haplotipos compartidos y la forma reticular que presenta el mapa de haplotipos de la región ITS2 y compartir la mayoría de los haplotipos de la región trnH-psbA. La capacidad de dispersión genética mediada por polinización anemófila, característica del género Quercus L., explica la existencia de flujo genético entre poblaciones separadas por grandes distancias, como se ha evidenciado en Q. robur L., donde se ha registrado polinización entre bosques aislados por hasta 30km (Ashley, 2021). Sin embargo, estudios en el norte de Nicaragua sobre Q. segoviensis Liebm. revelan estructuración genética significativa (FST ≥ 0,10) en poblaciones separadas por tan solo 20km, siendo la distancia geográfica el principal factor que explica esta diferenciación (Ortego et al., 2015). Nuestros resultados parecen representar un punto medio entre ambos casos, teniendo interconectividad genética entre los sitos Cerro Verde y Laguna de las Ninfas, separados por una distancia de 20km, y aislamiento en la población Volcán de San Vicente, separada por 80km. Si bien la prueba de Mantel no tiene significancia estadística (probablemente por el número reducido de sitios a comparar), nos sugiere que al igual que en estos estudios, la diferenciación genética se explique, al menos parcialmente, por la separación geográfica entre los grupos de individuos. Es importante considerar el contexto orográfico de los sitios analizados, siendo que los sitios Laguna de las Ninfas y Cerro Verde son parte de la cordillera Apaneca – Ilamatepec, en la que se encuentran múltiples parches boscosos naturales a distancias inferiores a 4km (Ministerio de Medio Ambiente y Recursos Naturales, 2004), mientras que la población presente en el Volcán de San Vicente se encuentra aislado de otras elevaciones de importancia (>1 000 m.s.n.m) por al menos 35km, esta información nos permite interpretar de una mejor manera los patrones de estructura genética presentes.
Los árboles filogenéticos nos muestran la existencia de dos linajes separados en Q. skinneri Benth., el primero, perteneciente a las poblaciones occidentales (Cerro Verde y Laguna de las Ninfas) y el segundo, compuesto por los individuos de la localidad Volcán de San Vicente, mostrándose el mismo patrón con ambos marcadores, pero con un mayor grado de diferenciación para el marcador ITS2, esto nuevamente concuerda con las características reproductivas propias del género Quercus L., con una capacidad de dispersión de semillas muy inferior a su capacidad de dispersión de polen. En su estudio en Q. laeta Liebm., una especie que presenta una extensa plasticidad morfológica, variando a lo largo de su distribución, (Morales-Saldaña et al., 2022) utilizó microsatélites nucleares y secuencias de espaciadores cloroplásticos para determinar la existencia de dos identidades específicas dentro de lo previamente definido como una única especie, si bien él estudió incluyó un mayor número de individuos distribuidos en una extensión geográfica más amplia, cabe resaltar la similitud entre sus resultados y los observados en nuestro estudio de Q. skinneri, por lo que se sugiere estudiar a más profundidad la situación de esta especie a nivel nacional y regional, ya que esto podría llevar a la reclasificación taxonómica de alguna de las poblaciones de esta especie.
En conclusión, nuestra investigación demuestra que regiones como ITS2 pueden ser herramientas útiles para el estudio de la diversidad genética, ya que en Q. skinneri los resultados obtenidos para la diversidad genética interespecífica fueron altamente informativos. Asimismo, identificamos una clara estructuración poblacional en Q. skinneri Benth., evidenciada tanto en el marcador nuclear como en el marcador cloroplástico, donde la población de San Vicente se diferencia del resto, lo que sugiere la posible existencia de linajes separados dentro de la especie.
Estos hallazgos amplían el conocimiento sobre la identificación de especies de robles en El Salvador. Para futuras investigaciones, se recomienda ampliar el muestreo, incluyendo un mayor número de individuos y poblaciones tanto a nivel nacional como regional. Además, para esclarecer la presencia de nuevos linajes o especies, se sugiere complementar los datos genéticos con información morfométrica. Este estudio contribuirá a generar un mayor conocimiento sobre la taxonomía y estructura poblacional del complejo Acutifolia, en el cual la identificación de especies representa un desafío.
AGRADECIMIENTOS
Agradecemos a la Facultad de Ciencias Naturales y Matemática de la Universidad de El Salvador por financiar el costo de secuenciación de este estudio. A Jenny Menjívar, encargada del Herbario del Museo de Historia Natural de El Salvador por su apertura y apoyo en la revisión de muestras, a Gabriel Cerén, de la Escuela de Biología de la Universidad de El Salvador, por su apoyo en la revisión de muestras y acompañamiento en el muestreo.
ÉTICA, CONFLICTO DE INTERESES Y DECLARACIÓN DE FINANCIAMIENTO
Declaramos haber cumplido con todos los requisitos éticos y legales pertinentes, tanto durante el estudio como en la preparación de este documento; que no hay conflictos de interés de ningún tipo, y que todas las fuentes financieras se detallan plena y claramente en la sección de agradecimientos. Asimismo, estamos de acuerdo con la versión editada final de esta publicación. El respectivo documento legal firmado se encuentra en los archivos de la revista. La declaración de contribución de cada autor es la siguiente: Todos los coautores: Diseño del estudio, recolección y análisis de datos, preparación y aprobación final del manuscrito.
REFERENCIAS
Ashley, M. V. (2021). Answers blowing in the wind: A quarter century of genetic studies of pollination in oaks. Forests, 12(5). https://doi.org/10.3390/f12050575
Aykut, Y. (2020). The importance in DNA barcoding of the regions which is covering rRNA genes and its sequences in the genus Quercus L. Bangladesh Journal of Plant Taxonomy, 27(2), 261–271. https://doi.org/10.3329/BJPT.V27I2.50666
Berendsohn, W. G., Gruber, A. K., & Monterrosa Salomón, J. (2009). Nova Silva Cuscatlanica. Árboles nativos e introducidos de El Salvador. Parte 1: Angiospermae – Familias A a L. BGBM.
Bolson, M., Smidt, E. de C., Brotto, M. L., & Silva-Pereira, V. (2015). ITS and trnH-psbA as Efficient DNA Barcodes to Identify Threatened Commercial Woody Angiosperms from Southern Brazilian Atlantic Rainforests. PLOS ONE, 10(12). https://doi.org/10.1371/journal.pone.0143049
Chen, S., Yao, H., Han, J., Liu, C., Song, J., Shi, L., Zhu, Y., Ma, X., Gao, T., Pang, X., Luo, K., Li, Y., Li, X., Jia, X., Lin, Y., & Leon, C. (2010). Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS ONE, 5(1). https://doi.org/10.1371/journal.pone.0008613
Dhivya, S., Ashutosh, S., Gowtham, I., Baskar, V., Harini, A. B., Mukunthakumar, S., & Sathishkumar, R. (2020). Molecular identification and evolutionary relationships between the subspecies of Musa by DNA barcodes. BMC genomics, 21(1), 659. https://doi.org/10.1186/S12864-020-07036-5
Durán Escalante, K. C., Ortiz Díaz, J. J., Pinzón Esquivel, J. P., & Gálvez Mariscal, M. A. (2023). Utilidad de los códigos de barras de DNA en la identificación de plantas melíferas asociadas a la miel monofloral de Sabal yapa producida en el este de Yucatán, México. https://doi.org/10.26461/26.01
Ersts, P. J. (s/f). Geographic Distance Matrix Generator (version 1.2.3). American Museum of Natural History, Center for Biodiversity and Conservation. Recuperado el 22 de febrero de 2025, de https://biodiversityinformatics.amnh.org/open_source/gdmg/index.html
Ewing, B., & Green, P. (1998). Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Research, 8(3), 186–194. https://doi.org/10.1101/gr.8.3.186
Excoffier, L., Laval, G., & Schneider, S. (2017). Arlequin (version 3.0): An integrated software package for population genetics data analysis: https://doi.org/10.1177/117693430500100003
Feng, S., Jiang, M., Shi, Y., Jiao, K., Shen, C., Lu, J., Ying, Q., & Wang, H. (2016). Application of the ribosomal DNA ITS2 region of physalis (Solanaceae): DNA barcoding and phylogenetic study. Frontiers in Plant Science, 7(2016JULY), 1047. https://doi.org/10.3389/FPLS.2016.01047
Feng, S., Jiang, Y., Wang, S., Jiang, M., Chen, Z., Ying, Q., & Wang, H. (2015). Molecular Identification of Dendrobium Species (Orchidaceae) Based on the DNA Barcode ITS2 Region and Its Application for Phylogenetic Study. International Journal of Molecular Sciences, 16(9), 21975–21988. https://doi.org/10.3390/ijms160921975
Fernández, J. F., Sork, V. L., Gallego, G., López, J., Bohorques, A., & Tohme, J. (2000). Cross-Amplification of Microsatellite Loci in a Neotropical Quercus Species and Standardization of DNA Extraction from Mature Leaves Dried in Silica Gel. Plant Molecular Biology Reporter, 18(4). https://doi.org/10.1007/BF02825070
Good, K., Coombes, A. J., Valencia-A, S., Rodríguez-Acosta, M., Bruns, E. B., & Alvarez-Clare, S. (2024). Análisis de Vacíos de Conservación de Especies Nativas de Encinos Mesoamericanos.
Gu, W., Song, J., Cao, Y., Sun, Q., Yao, H., Wu, Q., Chao, J., Zhou, J., Xue, W., & Duan, J. (2013). Application of the ITS2 Region for Barcoding Medicinal Plants of Selaginellaceae in Pteridophyta. PLoS ONE, 8(6). https://doi.org/10.1371/journal.pone.0067818
Hamilton. (1999). Four primer pairs for the amplification of chloroplast intergenic regions with intraspecific variation. Molecular ecology, 8(3).
Hebert, P. D. N., Cywinska, A., Ball, S. L., & DeWaard, J. R. (2003). Biological identifications through DNA barcodes. Proceedings of the Royal Society B: Biological Sciences, 270(1512), 313–321. https://doi.org/10.1098/rspb.2002.2218
Hipp, A. L., Manos, P. S., González-Rodríguez, A., Hahn, M., Kaproth, M., McVay, J. D., Avalos, S. V., & Cavender-Bares, J. (2018). Sympatric parallel diversification of major oak clades in the Americas and the origins of Mexican species diversity. New Phytologist, 217(1), 439–452. https://doi.org/10.1111/NPH.14773
Hipp, A. L., Manos, P. S., Hahn, M., Avishai, M., Bodénès, C., Cavender-Bares, J., Crowl, A. A., Deng, M., Denk, T., Fitz-Gibbon, S., Gailing, O., González-Elizondo, M. S., González-Rodríguez, A., Grimm, G. W., Jiang, X. L., Kremer, A., Lesur, I., McVay, J. D., Plomion, C., … Valencia-Avalos, S. (2020). Genomic landscape of the global oak phylogeny. New Phytologist, 226(4), 1198–1212. https://doi.org/10.1111/NPH.16162
Hollingsworth, P. M. (2011). Refining the DNA barcode for land plants. Proceedings of the National Academy of Sciences, 108(49), 19451–19452. https://doi.org/10.1073/PNAS.1116812108
Hollingsworth, P. M., Forrest, L. L., Spouge, J. L., Hajibabaei, M., Ratnasingham, S., van der Bank, M., Chase, M. W., Cowan, R. S., Erickson, D. L., Fazekas, A. J., Graham, S. W., James, K. E., Kim, K. J., John Kress, W., Schneider, H., van AlphenStahl, J., Barrett, S. C. H., van den Berg, C., Bogarin, D., … Little, D. P. (2009). A DNA barcode for land plants. Proceedings of the National Academy of Sciences of the United States of America, 106(31), 12794–12797. https://doi.org/10.1073/PNAS.0905845106
Hvilsom, C., Segelbacher, G., Ekblom, R., Fischer, M. C., Laikre, L., Leus, K., O’Brien, D., Shaw, R., & Sork, V. (2022). Selecting species and populations for monitoring of genetic diversity. IUCN, International Union for Conservation of Nature. https://doi.org/10.2305/IUCN.CH.2022.07.en
Jerome, D. (2020). Quercus skinneri. En IUCN Red List of Threatened Species 2020. https://doi.org/10.2305/IUCN.UK.2020-2.RLTS.T32768A2823212.en
Jump, A. S., Marchant, R., & Peñuelas, J. (2009). Environmental change and the option value of genetic diversity. Trends in Plant Science, 14(1), 51–58. https://doi.org/10.1016/J.TPLANTS.2008.10.002
Kress, W. J., Wurdack, K. J., Zimmer, E. A., Weigt, L. A., & Janzen, D. H. (2005). Use of DNA barcodes to identify flowering plants. Proceedings of the National Academy of Sciences of the United States of America, 102(23), 8369–8374. https://doi.org/10.1073/PNAS.0503123102
Kumar, S., Stecher, G., Suleski, M., Sanderford, M., Sharma, S., Tamura, K., & Ursula Battistuzzi, F. (2024). MEGA12: Molecular Evolutionary Genetic Analysis Version 12 for Adaptive and Green Computing. Molecular Biology and Evolution, 41(12), 1–9. https://doi.org/10.1093/MOLBEV/MSAE263
Lauer, W. (1954). Las formas de la vegetación de El Salvador (con 1 mapa). Comunicaciones, 3(1), 41–45.
Leigh, J. W., & Bryant, D. (2015). POPART: Full-feature software for haplotype network construction. Methods in Ecology and Evolution, 6(9). https://doi.org/10.1111/2041-210X.12410
Liu, H. Z., Takeichi, Y., Kamiya, K., & Harada, K. (2013). Phylogeography of Quercus phillyraeoides (Fagaceae) in Japan as revealed by chloroplast DNA variation. Journal of Forest Research, 18(4), 361–370. https://doi.org/10.1007/s10310-012-0357-y
Loera-Sánchez, M., Studer, B., & Kölliker, R. (2020). DNA barcode trnH-psbA is a promising candidate for efficient identification of forage legumes and grasses. BMC Research Notes, 13(1), 35. https://doi.org/10.1186/s13104-020-4897-5
Martins, K., Gugger, P. F., Llanderal-Mendoza, J., González-Rodríguez, A., Fitz-Gibbon, S. T., Zhao, J. L., Rodríguez-Correa, H., Oyama, K., & Sork, V. L. (2018). Landscape genomics provides evidence of climate-associated genetic variation in Mexican populations of Quercus rugosa. Evolutionary Applications, 11(10), 1842–1858. https://doi.org/10.1111/EVA.12684
Ministerio de Medio Ambiente y Recursos Naturales. (2004). Plan de manejo área natural Los Volcanes.
Morales-Saldaña, S., Valencia-Ávalos, S., Oyama, K., Tovar-Sánchez, E., Hipp, A. L., & González-Rodríguez, A. (2022). Even more oak species in Mexico? Genetic structure and morphological differentiation support the presence of at least two specific entities within Quercus laeta. JSE Journal of Systematics and Evolution. https://doi.org/10.1111/jse.12818
Okaura, T., Nguyen, D. Q., Ubukata, M., & Harada, K. (2007). Phylogeographic structure and late Quaternary population history of the Japanese oak Quercus mongolica var. crispula and related species revealed by chloroplast DNA variation. Genes and Genetic Systems, 82(6), 465–477. https://doi.org/10.1266/ggs.82.465
Ortego, J., Bonal, R., Muñoz, A., & Espelta, J. M. (2015). Living on the edge: The role of geography and environment in structuring genetic variation in the southernmost populations of a tropical oak. Plant Biology, 17(3), 676–683. https://doi.org/10.1111/plb.12272
Pacheco-Reyes, F. C., Wei, L., Pérez-Rodríguez, M. Á., Pacheco-Reyes, F. C., Wei, L., & Pérez-Rodríguez, M. Á. (2021). Análisis filogenético de especies de Quercus L. utilizando tres códigos de barras de ADN. Ecosistemas y recursos agropecuarios, 8(2). https://doi.org/10.19136/ERA.A8N2.2831
Pang, X., Song, J., Zhu, Y., Xu, H., Huang, L., & Chen, S. (2011). Applying plant DNA barcodes for Rosaceae species identification. Cladistics, 27(2), 165–170. https://doi.org/10.1111/J.1096-0031.2010.00328.X
Peakall, R., & Smouse, P. E. (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics, 28(19), 2537–2539. https://doi.org/10.1093/BIOINFORMATICS/BTS460
Qin, Y., Li, M., Cao, Y., Gao, Y., & Zhang, W. (2017). Molecular thresholds of ITS2 and their implications for molecular evolution and species identification in seed plants. Scientific Reports 2017 7:1, 7(1), 1–8. https://doi.org/10.1038/s41598-017-17695-2
Rodríguez-Acosta, M., & Coombes, A. J. (2020). Manual para la propagación de Quercus: Una guía fácil y rápida para cultivar encinos en México y América Central. 79.
Romero Rangel, S. (2006). Revisión taxonómica del complejo Acutifoliae de Quercus (fagaceae) con énfasis en su representación en México. Acta Botanica Mexicana, 76, 1–45.
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., & Sánchez-Gracia, A. (2017). DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Molecular Biology and Evolution, 34(12), 3299–3302. https://doi.org/10.1093/MOLBEV/MSX248
Simeone, M. C., Piredda, R., Papini, A., Vessella, F., & Schirone, B. (2013). Application of plastid and nuclear markers to DNA barcoding of Euro-Mediterranean oaks (Quercus, Fagaceae): Problems, prospects and phylogenetic implications. Botanical Journal of the Linnean Society, 172(4), 478–499. https://doi.org/10.1111/boj.12059
Sokołowska, J., Fuchs, H., & Celiński, K. (2022). Assessment of ITS2 Region Relevance for Taxa Discrimination and Phylogenetic Inference among Pinaceae. Plants (Basel, Switzerland), 11(8). https://doi.org/10.3390/PLANTS11081078
Valencia-A., S. (2017). Diversidad del género Quercus (Fagaceae) en México. Botanical Sciences, 75, 33. https://doi.org/10.17129/BOTSCI.1692
Valencia-Cuevas, L., Piñero, D., Mussali-Galante, P., Valencia-Ávalos, S., & Tovar-Sánchez, E. (2014). Effect of a red oak species gradient on genetic structure and diversity of Quercus Castanea (Fagaceae) in Mexico. Tree Genetics & Genomes, 10(3), 641–652. https://doi.org/10.1007/s11295-014-0710-8
White, T. J., Bruns, T., Lee, S., & Taylor, J. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. En PCR Protocols. https://doi.org/10.1016/b978-0-12-372180-8.50042-1
Xie, J., Chen, Y., Cai, G., Cai, R., Hu, Z., & Wang, H. (2023). Tree Visualization by One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Research, 51(W1). https://doi.org/10.1093/nar/gkad359
ANEXO 1
Tablas de Haplotipos.
TABLA A1
Haplotipos región ITS2
|
Posición |
85 |
115 |
139 |
151 |
152 |
155 |
156 |
186 |
189 |
221 |
239 |
250 |
288 |
|
Hap_1 |
C |
G |
T |
T |
C |
A |
T |
C |
C |
C |
C |
C |
A |
|
Hap_2 |
T |
. |
. |
. |
. |
. |
. |
. |
. |
T |
. |
T |
. |
|
Hap_3 |
. |
. |
. |
. |
. |
. |
. |
. |
. |
. |
. |
T |
. |
|
Hap_4 |
. |
. |
. |
. |
T |
G |
C |
. |
. |
. |
. |
T |
. |
|
Hap_5 |
T |
. |
. |
. |
T |
. |
. |
. |
. |
T |
. |
T |
. |
|
Hap_6 |
T |
. |
. |
C |
T |
G |
C |
T |
T |
. |
. |
T |
G |
|
Hap_7 |
T |
. |
. |
C |
T |
G |
C |
T |
. |
. |
. |
T |
G |
|
Hap_8 |
T |
. |
. |
C |
T |
G |
C |
T |
T |
. |
T |
T |
G |
|
Hap_9 |
T |
A |
A |
. |
. |
. |
. |
. |
. |
. |
. |
T |
. |
|
Hap_10 |
T |
. |
. |
. |
. |
. |
. |
. |
. |
. |
. |
. |
. |
TABLA A2
Haplotipos región trnH-psbA
|
Posición |
260 |
283 |
320 |
326 |
349 |
363 |
364 |
369 |
|
Hap_1 |
T |
A |
A |
A |
A |
G |
A |
T |
|
Hap_2 |
G |
C |
. |
. |
. |
T |
C |
. |
|
Hap_3 |
. |
. |
G |
G |
G |
. |
. |
G |