
UNED Research Journal (e-ISSN 1659-441X), Vol. 14(1): e3831, June, 2022
Makayla Tatiana Leza-Leza1![]() , Eunice Víquez-Ruiz2
, Eunice Víquez-Ruiz2![]() , Elías Barquero-Calvo3
, Elías Barquero-Calvo3![]() , Carolina Sancho-Blanco4
, Carolina Sancho-Blanco4![]() , Rodolfo Umaña-Castro4
, Rodolfo Umaña-Castro4![]()
1. Universidad Nacional de Costa Rica, Escuela de Ciencias Biológicas, Licenciatura en Biotecnología, Heredia, Costa Rica; makaylaleza@hotmail.com
2. Servicio Nacional de Salud Animal, Laboratorio de Bacteriología, Heredia, Costa Rica; eunice.viquez@senasa.go.cr,
3. Universidad Nacional de Costa Rica, Escuela de Medicina Veterinaria, Programa de Investigación de Enfermedades Tropicales, Heredia, Costa Rica; elias.barquero.calvo@una.ac.cr
4. Universidad Nacional de Costa Rica, Escuela de Ciencias Biológicas, Laboratorio de Análisis Genómico (LAGen), Heredia, Costa Rica; carolina.sancho.blanco@una.ac.cr, rodolfo.umana.castro@una.ac.cr
Recibido 05-XI-2021 Corregido 18-I-2022 Aceptado 21-II-2022
DOI: https://doi.org/10.22458/urj.v14i1.3831
|
ABSTRACT. "Optimization of PCR techniques optimization for detection of Salmonella enterica (serotype: Gallinarum) in Costa Rican poultry". Introduction: Avian typhoid and pullorosis, diseases caused by Salmonella enterica subsp. enterica (Gallinarum serotype, Gallinarum and Pullorum biotypes), cause high mortality in poultry and generate large economic losses. Objective: To optimize molecular techniques, such as endpoint PCR and real-time PCR (qPCR), to detect avian typhoid and pullorosis. Methods: We used control bacterial strains, isolates and tissues from birds infected with Salmonella Gallinarum to standardize detection with both techniques. Results: For the endpoint PCR, we obtained 100% repeatability, specificity and sensitivity, and a Kappa value of 0.98 for reproducibility; for qPCR, 103% efficiency with a variation under 6% in repeatability and reproducibility. The detection limit for genomic DNA was 6.4 pg/μL and the limit for the number of viable cells was 3x102 CFU/mL for endpoint PCR, and 10 DNA copies per reaction for qPCR. We also confirmed the identity of S. Gallinarum, and was reduced. We reduced the detection time to about 48 hours. Conclusion: We optimized a molecular technique for rapid, reliable and sensitive detection of Salmonella Gallinarum/Pullorum, which reduces the waiting time to take action in cases of clinical suspicion and possible outbreaks.
Keywords: molecular detection, DNA, taxonomic placement, Enterobacteriaceae
|
RESUMEN. Introducción: La tifosis aviar y la pulorosis, enfermedades ocasionadas por Salmonella enterica subsp. enterica (serotipo Gallinarum, biotipos Gallinarum y Pullorum), son responsables de una elevada mortalidad en aves de corral y generan grandes pérdidas económicas. Objetivo: Optimizar técnicas moleculares, como la PCR punto final y la PCR de tiempo real (qPCR), para detectar tifosis aviar y pulorosis. Métodos: Se emplearon cepas bacterianas control, aislamientos y tejidos de aves de infectadas con Salmonella Gallinarum para estandarizar la detección con ambas técnicas. Resultados: Para la PCR de punto final se obtuvo una repetibilidad, especificidad y sensibilidad del 100%, y un valor Kappa de 0,98 para la reproducibilidad; para qPCR una eficiencia del 103% y variación menor al 6% en repetibilidad y reproducibilidad. El límite de detección de ADN genómico fue de 6,4 pg/μL y el del número de células viables de 3x102 UFC/mL para la PCR punto final, y de 10 copias de ADN por reacción para la qPCR. Confirmamos la identidad de S. Gallinarum. Y se redujo el tiempo de detección a unas 48 horas, Conclusión: Logramos optimizar una técnica molecular para detección rápida, confiable y sensible de Salmonella Gallinarum/Pullorum, la cual reduce el tiempo de espera para tomar acción en casos de sospecha clínica y posibles brotes.
Palabras clave: detección molecular, ADN, posicionamiento taxonómico, Enterobacteriaceae.
|
Las bacterias pertenecientes al género Salmonella se caracterizan por ser Gram negativas, anaerobias facultativas y flageladas. Pertenecen a la familia Enterobacteriaceae y son un grupo muy complejo, presentando 6 subespecies con más de 2 500 serovariedades, dentro de las cuales se puede encontrar a Salmonella enterica subsp. enterica serotipo Gallinarum (Dalai et al. 2015). Este último serotipo presenta dos biotipos; Gallinarum (S. Gallinarum) y Pullorum (S. Pullorum); y a excepción del resto, no presenta motilidad debido a la ausencia de flagelos (Barrow & Freitas-Neto, 2011). Estos biotipos son responsables de producir la enfermedad sistémica conocida como tifosis aviar y pulorosis respectivamente, las cuales son exclusivas en aves, especialmente en gallinas de corral. Esto se debe a que dichos patógenos se encuentran muy adaptados a su hospedador, restringiéndose su transmisión a los seres humanos u otros animales (Foley et al., 2013). Ambas enfermedades causan, en la mayoría de los casos, una elevada mortalidad en las gallinas que se encuentran tanto en periodo de crecimiento como en adultos y genera, por lo tanto, grandes pérdidas económicas en la avicultura de diversos países (Barrow & Freitas-Neto, 2011). La detección de Salmonella se realiza principalmente mediante técnicas microbiológicas, sin embargo, en la última década se han realizado esfuerzos en cuanto a la búsqueda de técnicas más rápidas e igual de confiables, como lo es la PCR punto final y la PCR tiempo real (qPCR) (Xiong et al., 2016).
En Costa Rica la avicultura juega un papel muy importante en la economía del país al contribuir con un 13,6% en la producción pecuaria y, por lo tanto, generar miles de empleos a nivel nacional (Secretaría Ejecutiva de Planificación Sectorial Agropecuaria [SEPSA], 2020). Mediante el Decreto Ejecutivo No. 34669-MAG (2008) se establecen como enfermedades de declaración obligatoria en Costa Rica las ocasionadas por S. Enteritidis, S. Gallinarum y S. Pullorum. El Servicio Nacional de Salud Animal (SENASA) cuenta con un programa para la vigilancia, diagnóstico y tratamiento de enfermedades causadas por Salmonella spp. Sin embargo, no existe una norma que exija el uso de vacunas preventivas en las granjas; y la detección de la bacteria aún se realiza mediante una combinación de técnicas microbiológicas, inmunológicas y bioquímicas (Servicio Nacional de Salud Animal [SENASA], 2013). Estas técnicas tienen ciertas limitaciones asociadas a los tiempos de obtención de resultados, eficiencia, sensibilidad y especificidad. La detección empleando dichos métodos suele ser laboriosa y el tiempo aproximado para la obtención de resultados es entre 5 a 10 días (Beaubrun at al., 2017).
El presente trabajo tiene como objetivo facilitar la detección de los agentes causales de la tifosis aviar y la pulorosis mediante la optimización de técnicas moleculares, como la PCR punto final y la qPCR, utilizando cepas bacterianas control, aislamientos de aves de corral y tejidos infectados por Salmonella Gallinarum y Salmonella Pullorum.
MATERIALES Y MÉTODOS
Se optimizó la técnica de PCR punto final y qPCR para la detección del serotipo Gallinarum empleando muestras de cepas bacterianas y tejidos biológicos obtenidos a partir de aves de corral cedidas por el Laboratorio de Bacteriología del SENASA. Se cultivaron 4 aislamientos provenientes de un brote reportado en Costa Rica en el 2013, 8 cepas ATCC y 2 cepas vacunales (Apéndice Tabla S1) en caldo Tripticasa Soya (Acumedia®, Neogen) a 37°C durante 24 horas.
Extracción y cuantificación de ADN bacteriano: Se realizaron extracciones de ADN de las cepas aisladas y de los tejidos (hígado y médula ósea) de aves positivas para S. Gallinarum, empleando el kit de extracción DNeasy® Blood and Tissue (Qiagen, Alemania) y siguiendo el protocolo del fabricante. Los ácidos nucleicos aislados se almacenaron a -20°C para su posterior uso. El ADN genómico total se cuantificó mediante fluorimetría con el equipo Qubit 3.0 (Invitrogen™, Life Technologies) junto con el kit dsDNA HS (High Sensitivity, Invitrogen™, Life Technologies). La integridad del ADN se evaluó mediante electroforesis en gel de agarosa al 1% con tampón TBE (Tris-Borato) 0,5X y escalera de ADN GeneRuler 1Kb (Thermo Scientific). La pureza de las extracciones se analizó con un espectrofotómetro Nanodrop™ 2000 (Thermo Scientific).
Optimización de la PCR punto final y la qPCR: Se realizó la evaluación de la PCR punto final y los resultados se utilizaron para optimizar la qPCR. Para la PCR y sus variantes se emplearon los cebadores flhB (Fw 5’-TTC GCG ACG AAT TTA AAG AGA GCG AAG-3’) y flhB (Rev 5’CAG CGT TTA AGC TGC CAG ACC CAG GCC-3) propuestos por Xiong et al. (2016), los cuales están dirigidos a una región del gen flhB que codifica a la proteína de membrana biosintética flagelar (Flagellar biosynthetic protein). A pesar de que S. Gallinarum no es capaz de formar flagelos, presenta los genes estructurales conservados de flagelina. Los productos de PCR esperados fueron de 182pb para S. Gallinarum y de 379pb para otros serotipos de Salmonella spp. Como controles positivos se utilizaron la cepa vacunal SG 9R de S. Gallinarum (Nobilis®, Intervet International), la ATCC® 13036™ de S. Pullorum y otras cepas bacterianas (Apéndice Tabla S1).
Las reacciones PCR se realizaron a partir de 12,5μL de GoTaq® G2 Hot Start Green Master Mix (Promega) o EvaGreen® (type-it HRM PCR kit,Qiagen, Alemania) para PCR punto final y qPCR respectivamente, además de 0,33μL (0,4μM) de cada cebador (forward y reverse), 2μL de ADN y finalmente, se completó el volumen hasta 25,0μL con agua libre de nucleasas. El perfil térmico utilizado como base para la PCR punto final fue el siguiente: 95°C por 5 minutos; seguido de 30 ciclos a 95°C por 45 segundos, 59°C por 45 segundos, 72°C por 60 segundos y una extensión final a72°C durante 10 minutos. Mientras que, para la qPCR se eliminaron los pasos de 72°C por 60 segundos en el ciclo térmico y la extensión final. Se utilizó el equipo Mastercycler® Nexus Gradient (Eppendorf, Alemania) para la optimización con PCR punto final y el equipo QuantStudioTM Flex 6 (Applied Biosystems) para la qPCR. Para determinar la temperatura de hibridación óptima para los cebadores flhB-Fw y flhB-Rev, se realizó un gradiente de temperatura desde 56,0°C hasta 68,4°C utilizando la muestra control (cepa vacunal SG 9R) de S. Gallinarum.
La visualización de los productos de la PCR punto final se realizó mediante una electroforesis capilar con el equipo Qiaxcel Advanced (Qiagen) utilizando el cartucho QIAxcel DNA High Resolution, el marcador de peso molecular de 25-500pb, el marcador de alineamiento 15pb/600pb y el método de inyección OM800 (resolución 3-5pb). Las imágenes se obtuvieron con el QIAxcel ScreenGel, Software v.1.5.
La evaluación de la eficiencia (E) de la reacción mediante qPCR se realizó con una curva estándar empleando 4 diluciones seriadas 1:5 de la muestra control (cepa vacunal SG 9R), partiendo con una concentración inicial de 20ng/μL. Por lo tanto, la curva se construyó con un total de 5 puntos, cada uno por triplicado. Mientras que, la especificidad de los cebadores se evalúo mediante una curva de disociación utilizando la misma muestra control y la cepa ATCC 13076 de Salmonella Enteritidis.
Secuenciación del producto de PCR y análisis de posicionamiento taxonómico: Los productos de PCR obtenidos se purificaron mediante una precipitación con isopropanol/acetato de sodio. Para confirmar la identidad de las amplificaciones se realizó una secuenciación Sanger en dos direcciones (sentido y antisentido) de una región parcial del gen flhB, de los aislados de tres cepas de S. Gallinarum (cepa 2, cepa 4 y cepa 5), dos de S. Enteritidis (cepa 6 y cepa 9), una de S. Typhimurium (cepa 10) y una de S. Pullorum (cepa 11) (Apéndice Tabla S1), utilizando la química Big Dye terminator Cycle Sequencing v3.1 y posteriormente purificados mediante el kit Big Dye Xterminator Purification (Applied Biosystems), siguiendo las recomendaciones del fabricante. Finalmente, la identificación nucleotídica se realizó en un Analizador Genético ABI 3500 (Applied Biosystems) ubicado en el Laboratorio de Análisis Genómico (LAGen) de la Escuela de Ciencias Biológicas, Universidad Nacional, Costa Rica.
Para el análisis de posicionamiento taxonómico, las secuencias obtenidas se editaron manualmente en el programa Geneious versión R9 (Biomatters). El reconocimiento preliminar de su identidad taxonómica a nivel de género se realizó en línea con el programa BLASTn (Chen et al., 2015), mediante parámetros por defecto y disponible en National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/blast). Se llevó a cabo un alineamiento múltiple en el programa MAFFT v7.308 (Katoh & Standley, 2013) empleando el método iterativo de refinamiento G-INS-i (1PAM/k= 2), además, una selección de bloques conservados mediante los parámetros por defecto del programa en línea Gblocks (Castresana, 2000). Se determinó el modelo de sustitución nucleotídica para los bloques conservados de la región génica flhB (181 posiciones informativas) con el programa ModelFinder (Kalyaanamoorthy et al., 2017) indicando que el mejor modelo ajustado a la matriz fue el K2P. Seguidamente, con la plataforma en línea IQ-TREE (http://iqtree.cibiv.univie.ac.at) (Nguyen et al., 2015; Trifinopoulos et al., 2016) se obtuvieron las inferencias topológicas de posicionamiento taxonómico por máxima verosimilitud con el uso de un algoritmo estocástico. Los parámetros empleados para la inferencia filogenética fueron el uso de heterogeneidad de tasa libre, cálculo de soporte de los nodos derivado del método bootstrap UFboot con 5000 permutaciones (Minh et al., 2013) y un test de rama única (SH-aLRT) con 5000 réplicas. Finalmente, los árboles obtenidos se visualizaron y editaron en el programa FigTree v1.4 (Rambaut 2009).
Análisis de especificidad, sensibilidad y límite de detección de la PCR: La especificidad y sensibilidad se evaluó mediante la amplificación por PCR punto final tanto de cepas de Salmonella spp. como de otros géneros (Apéndice Tabla S1), validándose un total de 28 reacciones de PCR. La cepa vacunal SG 9R de S. Gallinarum se empleó como control positivo. Ambos parámetros se calcularon con las ecuaciones descritas por Trevethan (2017).
Para determinar el menor número de células que pueden ser detectadas mediante PCR punto final, la cepa vacunal SG 9R fue ajustada a una concentración aproximada de 1.5x108 UFC/mL correspondiente al patrón 0,5 de McFarland y a partir de dicha suspensión se realizaron diluciones seriadas 1:10 desde 10-1 hasta 10-8 en caldo Tripticasa Soya. Luego se sembraron 0,01mL de cada dilución por triplicado en diferentes placas Petri con Agar Tripticasa Soya y se incubaron durante 24 horas a 37°C. Posteriormente se realizó un recuento en placa de las unidades formadoras de colonias (UFC) y un análisis por PCR punto final a partir de las colonias obtenidas en cada una de las placas. De manera similar, se evaluó la concentración más baja de ADN genómico para determinar el límite de detección de S. Gallinarum mediante PCR punto final realizando diluciones seriadas 1:5 del ADN de la cepa vacunal SG 9R, partiendo de una concentración de 20ng/μL y hasta 1,28pg/μL. A partir de los resultados obtenidos de la PCR punto final, se llevaron a cabo diluciones seriadas 1:10 desde 10-1 hasta 10-10 de la cepa vacunal SG 9R para determinar la sensibilidad analítica de la detección mediante qPCR. Se ensayaron únicamente las diluciones de 10-7, 10-8, 10-9 y 10-10. El valor de Ct establecido como corte para considerar el resultado como positivo fue de 35 (World Organization for Animal Health, 2014).
Análisis de repetibilidad y reproducibilidad: Ambas técnicas (PCR punto final y qPCR) se validaron mediante una prueba de repetibilidad (análisis intralaboratorio) en el Laboratorio de Bacteriología del SENASA, utilizando los mismos materiales y condiciones de laboratorio durante cada ensayo. Para la PCR punto final se utilizaron 3 cepas de S. Gallinarum (cepa 1, cepa 5 y cepa 11), una de S. Abaetetuba (cepa 8) y una de S. Enteritidis (cepa 9), en este caso se emplearon 10 réplicas de cada una de las muestras y se analizó la concordancia entre los resultados de las réplicas. Para la qPCR se llevó a cabo la amplificación de 5 diluciones 1:5 de la cepa vacunal SG 9R la cual se encontraba a una concentración inicial de 20ng/μL.
La reproducibilidad (análisis interlaboratorio) de la PCR tiempo final y qPCR se analizó comparando los resultados de repetibilidad obtenidos en diferentes laboratorios, validando así la prueba bajo condiciones experimentales diferentes. Para la PCR punto final se realizaron comparaciones entre el Laboratorio de Bacteriología del SENASA y el LAGen, utilizando los equipos Mastercycler® Nexus Gradient (Eppendorf, Alemania) del SENASA y equipo ProFlex™ PCR System (Applied Biosystems, USA) del LAGen. Mientras que, para la qPCR se compararon los resultados entre el Laboratorio de Bacteriología del SENASA y el Laboratorio de Biotecnología de Plantas (LBP) de la UNA, con los termocicladores QuantStudio™ Flex 6 (Applied Biosystems) y LightCycler® 96 Instrument (Roche) del SENASA y el LBP respectivamente.
Análisis estadístico de la reproducibilidad y repetibilidad: Para determinar la reproducibilidad de la técnica de PCR punto final se elaboró una tabla de contingencia 2X2 comparando los resultados del SENASA (Laboratorio 1) con los del LAGen (Laboratorio 2). Con los datos obtenidos se calculó el índice de Kappa para determinar el grado de concordancia entre los diferentes días y se realizó la prueba de Mc Nemar. Se determinó si los datos eran estadísticamente similares mediante la comparación del resultado de la prueba de Mc Nemar con un valor tabular de Chi Cuadrado de X2Chi= 3,84, utilizando como criterio de decisión un nivel de significancia de 0,05 y un grado de libertad (Díaz et al., 2017).
La repetibilidad de la prueba mediante qPCR se obtuvo calculando el valor de la desviación estándar (DS) y el porcentaje del coeficiente de variación (% CV) para las siguientes concentraciones por triplicado: 20ng/μL, 4ng/μL, 0,8ng/μL, 0,16ng/μL, 0,032ng/μL y 0,0064ng/μL de la cepa control (SG 9R) dentro de un mismo ensayo en el termociclador QuantStudio™ Flex 6 (Applied Biosystems). Mientras que, para calcular la reproducibilidad de la misma prueba se compararon los datos de DS y del % CV obtenidos en dos laboratorios diferentes (Laboratorio de Bacteriología del SENASA y el LAGen) utilizando las concentraciones mencionadas.
Detección de S. Gallinarum/Pullorum en tejidos biológicos: Con el fin de evaluar la aplicabilidad de la técnica de la PCR para detectar S. Gallinarum/Pullorum directamente en muestras de tejido biológico sin requerir de un aislamiento y obtención de un cultivo puro, se realizó una extracción de ADN genómico directamente de muestras congeladas de hígado y médula ósea de diferentes animales previamente confirmadas como positivas para S. Gallinarum/Pullorum mediante cultivo bacteriano, pruebas serológicas y bioquímicas (com pers. Gabriela Hernández, SENASA, octubre 2019) y posteriormente se llevó a cabo un análisis por PCR en tiempo final con las condiciones previamente optimizadas.
RESULTADOS
Extracción y cuantificación de ADN bacteriano: La concentración de ADN total obtenido varió entre las muestras, en un rango entre 14ng/μL a 46,8ng/μL. En la evaluación de la integridad del ADN extraído mediante electroforesis no se observó degradación y se muestran bandas de alto peso molecular (Apéndice figura S1). Al analizar la pureza de las extracciones mediante la relación de absorbancia 260/280 (A260/280) se obtuvieron valores cercanos a 1,8 mientras que, en la relación de A260/230 se encontraron por debajo de 1,8.
Optimización de la PCR punto final y la qPCR: Al emplear el gradiente de temperatura se observó una amplificación positiva desde 56℃ hasta 68,4℃ (Figura 1). Sin embargo, al analizar los productos obtenidos para cada una de las muestras con el programa QIAxcel ScreenGel, se evidenció una mayor concentración del producto de interés (182pb) y menos artefactos de la PCR al emplear una temperatura entre 57,1℃ a 59,9℃ (datos no mostrados) por lo tanto, se optó por utilizar una temperatura de 59,0℃ durante la etapa de hibridación con el fin de mantener una alta especificidad durante las pruebas.
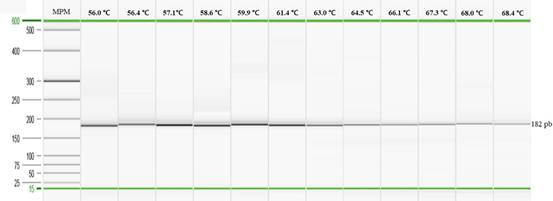
Fig. 1. Electroforesis capilar para la determinación de la temperatura de hibridación empleando la cepa de S. Gallinarum (vacunal SG 9R).
La eficiencia obtenida a partir de la curva estándar fue de E= 103%, con un R2 de 0,998. Los valores de Ct se mantuvieron entre 13.633 y 22.614 (figura 2 y 3).
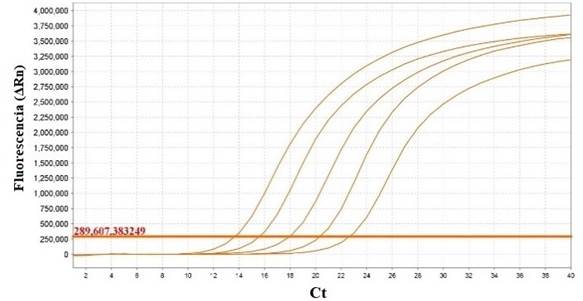
Fig. 2. Curvas de amplificación de las diluciones seriadas del ADN de la cepa vacunal SG 9R
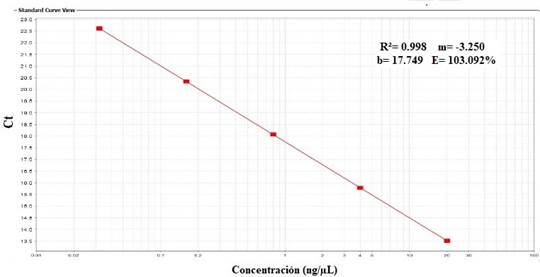
Fig. 3. Curva estándar obtenida a partir de las diluciones seriadas de la cepa vacunal SG 9R
En cuanto a la curva de disociación obtenida para ambos serotipos de Salmonella spp. se observó un único pico con temperaturas diferentes para cada uno de los productos de PCR (182pb y 379pb) (Figura 4).
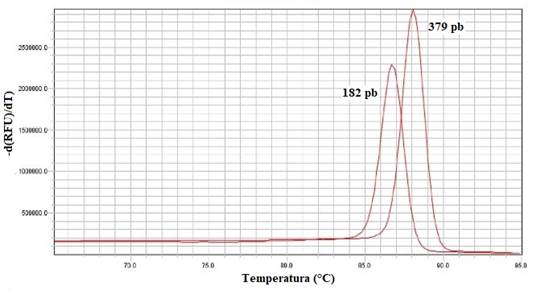
Fig. 4. Curvas de disociación obtenidas mediante qPCR para S. Gallinarum (182pb) y S. Enteritidis (379pb)
Análisis de posicionamiento taxonómico: La identidad molecular de las muestras secuenciadas se comprobó mediante una inferencia filogenética (Figura 5) y (Apéndice Tabla S2). Las cepas de S. Gallinarum/Pullorum se agruparon junto con cepas reportadas en el NCBI que pertenecían a la misma especie, lo mismo ocurrió con las cepas de serotipos diferentes al de S. Gallinarum. El valor de soporte para las ramas fue de 81/64.
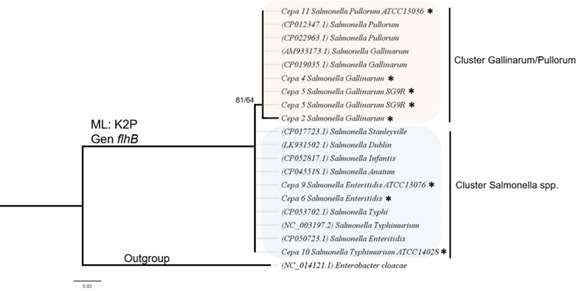
Fig. 5. Árbol de posicionamiento taxonómico basado en el método de máxima verosimilitud (ML) de los fragmentos del gen flhB de cepas utilizadas en este estudio (*). El soporte de cada una de las ramas se muestra como SHaLRT (Test de rama única) /UFBoot (Bootstrap ultra rápido). En cada rama se muestra el número de accesión GenBank utilizado para el análisis. La especie Enterobacter cloacae se utilizó como grupo externo.
Estudio de especificidad y sensibilidad en la detección por PCR: Se obtuvo una especificidad y una sensibilidad del 100% para la PCR punto final al emplear un total de 14 cepas diferentes (Figura 6)
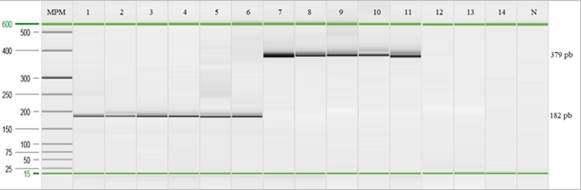
Fig. 6. Electroforesis capilar para la determinación de la especificidad y sensibilidad.; de izquierda a derecha: (MPM) marcador molecular (25pb a 500pb); ADN de S. Gallinarum cepa 1, 2, 3 ,4 ,5 y 11; S. Enteritidis cepa 9 y 6; S. Abaetetuba cepa 8; S. Choleraesuis cepa 7; S. Typhimurium cepa 10; P. multocida cepa 14; S. aureus cepa 12; E. coli cepa 13; (N) control negativo (agua doble desionizada libre de nucleasas).
Límite de detección (sensibilidad analítica): Se logró observar la formación de colonias hasta la dilución 10-6 y al llevar a cabo la PCR punto final de cada dilución se obtiene un límite de detección para células bacterianas viables de 3x102UFC/mL (Figura 7A). Mientras que, el límite de detección para el ADN genómico mediante PCR punto final fue de 6,4pg/μL (Figura 7B).
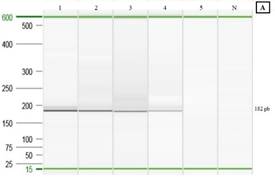

Fig. 7. Electroforesis capilar para la determinación del límite de detección de células bacterianas viables y ADN genómico. A: (1) 1x105UFC/mL; (2) 1x104UFC/mL; (3) 2x103UFC/mL; (4) 3x102UFC/mL; (5) 1x102UFC/mL; (N) control negativo. B: (1) 20ng/μL; (2) 4ng/μL; (3) 800pg/μL; (4) 160pg/μL; (5) 32pg/μL; (6) 6,4pg/μL; (7) 1,28pg/μL; (N) Control negativo.
El límite de detección obtenido para la qPCR fue de 2X10-9 ng/μL (dilución de 10-9) con un valor de Ct de 34.501 ± 0,133 lo cual equivale a 10 copias de ADN por reacción (Figura 8).
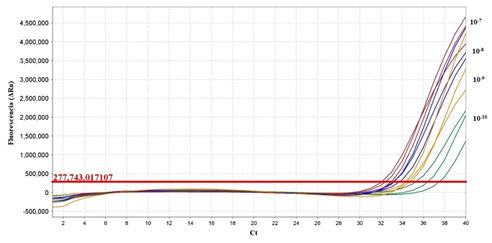
Fig. 8. Curva de amplificación para el límite de detección mediante qPCR de las diluciones seriadas de 10-7, 10-8, 10-9 y
10-10 del ADN genómico de la cepa vacunal SG 9R.
Análisis de reproducibilidad y repetibilidad: Se obtuvo un 100% de repetibilidad para la PCR punto final al comparar las réplicas dentro del mismo ensayo y para el análisis de reproducibilidad se obtuvo un valor de Kappa de K= 0,98 lo cual corresponde a un grado de concordancia “excelente” (Díaz et al., 2017).
Al aplicar la prueba estadística de Mc Nemar, a partir de los resultados obtenidos de la tabla de contingencia 2x2, se obtuvo un X2Mc= 1,00 y al realizar la comparación con el valor Chi Xchi = 3,84 se concluyó que X2Mc<Xchi por lo tanto, se aceptó la hipótesis Nula (Ho) y se estableció que no existen diferencias estadísticamente significativas entre los resultados del Laboratorio 1 y el Laboratorio 2.
Para la qPCR en el ensayo intra-laboratorio se logró evidenciar una elevada repetibilidad debido a los valores bajos (≤0,102) que se obtuvieron para la DS y % CV (≤0,673) entre cada una de las réplicas (Tabla 1). De igual manera, los resultados obtenidos para el análisis inter-laboratorio se mantuvieron entre 0,285 a 1,088 para la DS y 1,225 a 5,904 en el caso del % CV, demostrando una alta reproducibilidad de la prueba al realizar la comparación entre ambos laboratorios (Tabla 2).
Ensayo intra-laboratorio de la qPCR
|
Muestra (ng/μL) |
Promedio Ct (n=3) a |
DS b |
CV (%)c |
|
20 |
14,407 ± 0,020 |
0,035 |
0,244 |
|
4 |
16,400 ± 0,050 |
0,087 |
0,532 |
|
0,8 |
19,197 ± 0,012 |
0,021 |
0,108 |
|
0,16 |
22,280 ± 0,086 |
0,150 |
0,673 |
|
0,032 |
23,057 ± 0,058 |
0,102 |
0,443 |
|
0,0064 |
24,907 ± 0,044 |
0,076 |
0,307 |
a Valores de la media de Ct ± error estándar de la media para las 3 réplicas (n=3)
b Desviación Estándar
c Porcentaje del Coeficiente de Variación
Ensayo inter-laboratorio de la qPCR
|
Muestra (ng/μL) |
Promedio Ct a |
DS b |
CV (%)c |
|
|
SENASA |
LBP |
|||
|
20 |
13,680 ± 0,156 |
14,407 ± 0,020 |
0,514 |
3,657 |
|
4 |
15,373 ± 0,161 |
16,400 ± 0,050 |
0,726 |
4,571 |
|
0,8 |
17,658 ± 0,069 |
19,197 ± 0,012 |
1,088 |
5,904 |
|
0,16 |
21,493 ± 0,017 |
22,280 ± 0,086 |
0,557 |
2,544 |
|
0,032 |
23,460 ± 0,132 |
23,057 ± 0,058 |
0,285 |
1,225 |
|
0,0064 |
25,775 ± 0,046 |
24,907 ± 0,044 |
0,614 |
2,422 |
a Valores de la media de Ct ± error estándar de la media para las 3 réplicas (n=3)
b Desviación Estándar
c Porcentaje del Coeficiente de Variación
LBP: Laboratorio de Biotecnología de Plantas
Detección de S. Gallinarum/Pullorum en tejidos biológicos: En los cultivos de los tejidos positivos para S. Gallinarum/Pullorum se observaron muy pocas colonias de Salmonella. En el caso del cultivo correspondiente a la médula ósea no se obtuvo ninguna colonia. A pesar de las pocas colonias de S. Gallinarum en los cultivos en placa, al realizar la PCR punto final directamente de los tejidos biológicos se lograron obtener productos PCR positivos para cada uno de los tejidos en estudio (Figura 9). Cabe mencionar que, el tiempo para la obtención de resultados con cada una de las técnicas de PCR fue de aproximadamente 48 horas.
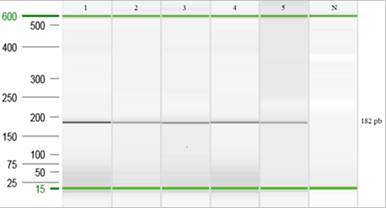
Fig. 9. Electroforesis capilar de las muestras de tejido biológico. (1) Control positivo (cepa SG 9R); (2 y 3) Hígado; (4 y 5) Médula ósea; (N) control negativo (agua doble desionizada libre de nucleasas).
DISCUSIÓN
El procedimiento de extracción de ácidos nucleicos que se realizó en este estudio muestra una recuperación de ADN total adecuado para posteriores análisis de detección molecular. Sin embargo, las concentraciones de ADN obtenidas fueron menores a las reportadas por autores como Nurjanah et al. (2018), los cuales, mediante diferentes protocolos, lograron extraer hasta 200ng/μl de ADN a partir de cultivos de S. Typhimurium y S. Enteritidis. Por otro lado, al evaluar la pureza del ADN extraído en la relación de absorbancia 260/280 (A260/280) se obtuvieron valores cercanos a 1,8 lo cual refleja una baja contaminación con proteínas. Mientras que, los valores por debajo de 1,8 obtenidos para la relación de A260/230 podrían deberse a una posible contaminación con carbohidratos, fenol o sales (Lorenz, 2012). Según Koetsier y Cantor (2019) esto ocurre al utilizar una técnica de extracción basada en columnas, ya que algunos de los buffers utilizados contienen hidrocloruro de guanidina, la cual absorbe a dichas longitudes de onda. No obstante, las reacciones de PCR no mostraron inhibición por los contaminantes presentes en las extracciones y fue posible amplificar cada una de las muestras de este estudio.
El porcentaje de eficiencia obtenido para la qPCR demuestra que el producto de la amplificación se duplica en cada uno de los ciclos hasta completar los 40 ciclos. Según Nurjanah et al. (2018) el porcentaje de eficiencia aceptable para una qPCR se debe encontrar entre 90% y 110% con una pendiente de -3,6 a -3,1. Además, un coeficiente de regresión (R2) cercano a 1,0, como el obtenido, corresponde a una elevada correlación entre los valores de Ct y la cantidad de ADN (Álvarez et al., 2018) demostrando que, durante la etapa exponencial de la reacción existe una duplicación del ADN molde luego de cada ciclo, permitiendo así obtener una elevada eficiencia. Dichos resultados se pueden comparar con los obtenidos por Heymans et al. (2018), los cuales reportaron una eficiencia de 103,8% para la detección de Salmonella Typhimurium y Salmonella Enteritidis mediante qPCR. Lo anterior, se evidencia mediante la curva de disociación que no mostró la formación de dímeros de cebadores, estos últimos fueron altamente específicos para la amplificación de la región parcial del gen flhB.
El posicionamiento taxonómico de las amplificaciones por PCR permitió confirmar la identidad de las cepas empleadas en este estudio, corroborando la especificidad de los cebadores para amplificar el gen de interés. Liu et al. (2017) observaron una agrupación similar entre los diferentes serotipos de Salmonella spp. al analizar los genes de flagelina (flhB). En dicho estudio los serotipos S. Gallinarum, S. Enteritidis, S. Typhi, S. Stanleyville, S. Dublin y S. Typhimurium se posicionaron en un mismo clúster correspondiente a las especies monofásicas, mientras que S. Infantis y S. Anatum se agruparon con las especies difásicas. Sin embargo, debido a que todas pertenecen a la misma subespecie, la distancia entre grupos fue corta. Por otro lado, los cebadores utilizados en el presente estudio amplifican un fragmento pequeño (182 pb) de Salmonella spp. lo cual puede afectar la capacidad de resolución para diferenciar entre los serotipos de la especie debido al bajo número de posiciones informativas obtenidas de este producto de PCR.
Los valores de especificidad y sensibilidad obtenidos son similares a los reportados por Lavalett et al. (2009) durante la validación de una PCR múltiple. Estos reportaron valores entre 98% y 100% para la sensibilidad y entre 96% y 100% para la especificidad al emplear dos cebadores diferentes para la detección de 4 serotipos de Salmonella spp. de importancia clínica. Por otro lado, Xiong et al. (2016) obtuvo un valor del 100% para ambos parámetros empleando 27 serotipos diferentes de Salmonella spp. utilizando el mismo cebador (flhB). Según la World Organization for Animal Health (2019), el valor de dichos parámetros debe ser cercano al 99 % si se utilizan un número de muestras igual o menor a 15. Por lo tanto, nuestros resultados demuestran la capacidad de los cebadores utilizados de amplificar de manera eficiente el gen de interés en diferentes serotipos de Salmonella spp.
Los análisis del límite de detección para células o ADN permiten valorar la capacidad que tiene la técnica de PCR en detectar concentraciones muy bajas del analito, contribuyendo en una detección más precisa en muestras con una baja carga del patógeno. En cuanto al límite de detección para células bacterianas viables mediante PCR punto final, este concuerda con el reportado por Xiong et al. (2018) para la detección de S. Gallinarum/Pullorum. En dicho estudio, se obtuvo un límite de detección de 1x102 UFC/mL empleando cebadores dirigidos al gen ratA. De manera similar, el límite para ADN genómico se aproxima al obtenido por Xiong et al. (2016), quienes reportaron un valor de 5,85pg/μL empleando el mismo cebador. Mientras que, el límite de detección para la qPCR es el mismo que el reportado por Braun y Methner (2011) al validar la qPCR para la detección de Salmonella spp., afirmando que este valor cumple con los estándares internacionales según ISO 6579:2002, Anexo D.
El valor de la repetibilidad para la PCR punto final coincide con lo obtenido por Díaz de la Osa et al. (2015) durante la estandarización de una PCR punto final para la detección de potyvirus. Además, el grado de concordancia de Kappa (K), obtenido a partir de dicho análisis, es comparable con el reportado por Lavalett et al. (2009), quienes obtuvieron un K= 0,95 para la detección de Salmonella entérica, por lo que, es posible afirmar la reproducibilidad de la PCR punto final del presente estudio. En cuanto a la qPCR, el coeficiente de variación (CV) corresponde a la dispersión de los datos con respecto a la media calculada (Kralik & Ricchi, 2017), este coeficiente podría emplearse para evaluar la repetibilidad y reproducibilidad de un ensayo, ya que entre más bajo sea el %CV menor será la variabilidad entre los datos. En nuestro caso, los valores obtenidos para el coeficiente de variación demuestran una elevada repetibilidad y reproducibilidad de la qPCR, de manera similar comparables con valores obtenidos por otros autores (Ebentier et al., 2013; García, 2019), quienes reportaron valores de CV menores al 6%, reflejando la robustez y la fiabilidad en la generación del mismo resultado en diferentes ensayos.
En relación con el crecimiento reducido de las colonias durante el cultivo de tejidos, este se puede deber a que las muestras se almacenaron durante un período de tiempo prolongado (superior a 1 año) y, además, se sometieron a procesos de congelación y descongelación sin el uso de un agente crioprotector. Lo anterior puede ocasionar una reducción en la viabilidad celular mediante la ruptura de la membrana plasmática causada por la formación de cristales durante el proceso de congelación o al producir un choque osmótico cuando se descongela (Pegg, 2015). Sin embargo, los resultados de la PCR demostraron que no es necesario realizar una incubación previa en un medio nutritivo tal como Tripticasa soya o llevar a cabo un aislamiento del patógeno para lograr amplificar la secuencia de interés. Lo anterior es relevante para la rápida detección del patógeno en muestras obtenidas directamente de campo, previo a cualquier procedimiento de cultivo y aislamiento de la bacteria.
Las técnicas de PCR empleadas, permiten una detección más rápida al compararlas con las pruebas microbiológicas tradicionales, aportando una herramienta útil para disminuir el tiempo de espera para tomar acción en casos de sospecha clínica y posibles brotes. Además, mediante la secuenciación y posterior análisis de posicionamiento taxonómico se corrobora la amplificación exitosa y la identidad molecular del gen flhB correspondiente a S. Gallinarum/Pullorum.
Tanto la PCR tiempo final como la PCR tiempo real son capaces de detectar concentraciones muy bajas de S. Gallinarum/Pullorum. En este estudio, el cambio en las condiciones y de los equipos de laboratorio no afectó los resultados, demostrando la consistencia del método en producir los mismos resultados esperados. Además, se reduce el tiempo de espera para la obtención de resultados, lo cual contribuye en la pronta toma de decisiones, tales como el aislamiento o cuarentena de granjas y/o sacrificio de aves infectadas. Es importante mencionar que, aunque estos ensayos de biología molecular representan un costo mayor que las pruebas microbiológicas tradicionales (aproximadamente 40% más costosos), no se requiere de repeticiones o un gran número de análisis para confirmar la presencia del patógeno y al ser estas de importancia para la salud animal, el Servicio Nacional de Salud Animal (SENASA) exonera los costos económicos a los productores con respecto a estos análisis diagnósticos, ante sospechas de la presencia de esta enfermedad.
Con el fin de establecer con mayor robustez la especificidad y la selectividad de la PCR de detección de Salmonella que afecta granjas avícolas, es necesario analizar un mayor número de muestras de tejido biológico y diferentes serotipos de S. Gallinarum. Además, se debe procurar un almacenamiento adecuado de dichos tejidos junto con un agente crioprotector para evitar la posible degradación del ADN y lograr preservarlo por más tiempo previo al análisis molecular, esto debido a las pocas colonias bacterianas reportadas para las muestras de tejido biológico.
Otro aspecto importante que se debe considerar para futuros ensayos es el uso de cebadores que permitan diferenciar entre los biotipos Gallinarum y Pullorum con el fin de conocer la prevalencia de cada uno de estos en el país y utilizar dichos resultados para contribuir en el control y prevención de las enfermedades que estos biotipos ocasionan.
Finalmente, la identificación del patógeno directamente de tejido especifico de aves de corral fue exitosa, pero requiere de un mayor número de análisis a futuro que permitan establecer protocolos óptimos de detección, lo cual permitiría a la entidad encargada del monitoreo de patógenos en fincas avícolas, establecer menores tiempos de espera y respuesta en la toma de decisiones en casos de sospecha clínica y eventuales brotes. En el presente trabajo, se optimizaron dos métodos de detección por PCR, los cuales se muestran sensibles, confiables y rápidos para la detección de S. Gallinarum/Pullorum en aislamientos bacterianos obtenidos a partir de aves de corral.
AGRADECIMIENTOS
Agradecemos al Servicio Nacional de Salud Animal (SENASA) por permitir la ejecución de este trabajo, en especial por el acceso a recursos, banco de aislamientos y tejidos en sus instalaciones. Al Laboratorio de Biotecnología de Plantas (LBP) de la Escuela de Ciencias Biológicas, UNA, por facilitar el equipo LightCycler® 96 Instrument (Roche). A la Vicerrectoría de Investigación de la Universidad Nacional por el fondo económico brindado a través del Fondo para el Fortalecimiento de las Capacidades Estudiantiles en Investigación (FOCAES).
ÉTICA, CONFLICTO DE INTERESES Y DECLARACIÓN DE FINANCIAMIENTO
Los autores declaran que han cumplido plenamente con todos los requisitos éticos y legales pertinentes, tanto durante el estudio como en la producción del manuscrito; que no existen conflictos de interés de ningún tipo; que todas las fuentes financieras se detallan plena y claramente en la sección de agradecimientos; y están de acuerdo con la versión final editada del artículo. El documento legal firmado se encuentra en los archivos de la revista.
La contribución de cada autor al manuscrito es la siguiente: M.T.L.: análisis microbiológicos y moleculares, análisis de datos y redacción del manuscrito científico. Co-autores: análisis microbiológicos y moleculares, análisis de datos, preparación y aprobación final del manuscrito.
REFERENCIAS
Álvarez, O., Larigauderie, G., Ortega, G. M., Granja, A. C., & Cabria, R. J. (2018). Optimización y Validación de una PCR en Tiempo Real para la Rápida Identificación de Bacillus thuringiensis, Simulador de Bacillus anthracis. Sanidad Militar, 74(2), 84-89.
Barrow, P. A., & Freitas-Neto, O. C. (2011). Pullorum disease and fowl typhoid-new thoughts on old diseases: A review. Avian Pathology, 40(1), 1–13. https://doi.org/10.1080/03079457.2010.542575
Beaubrun, J. J. G., Ewing, L., Dudley, K., Benhamed, F., Wang, H., & Hanes, D. E. (2017). Evaluation of a multiplex PCR method to serotype Salmonella in animal feeds pre-enrichment broth cultures. MethodsX, 4(1), 335-345. https://doi.org/10.1016/j.mex.2017.09.003
Braun, S. D., & Methner, U. (2011). Comparison of DNA isolation methods and detection of Salmonella spp. from animal faeces and dust using invA real- time PCR. Berliner Und Munchener Tierarztliche Wochenschrift, 124(6), 177–185. https://doi.org/10.2376/0005-9366-124-177.
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular biology and evolution, 17(4), 540-552.
Chen, Y., Ye, W., Zhang, Y., & Xu, Y. (2015). High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Research, 43(16), 7762–7768. https://doi.org/10.1093/nar/gkv784
Dalai, N., Shekhar, S., Padhy, A., Praveen, P. K., & Sahu, A. R. (2015). Salmonellosis - a potential threat to poultry. Journal of Cell and Tissue Research, 15(3), 5209–5213.
Decreto Ejecutivo No. 34669-MAG (13 de agosto de 2008). Listado de enfermedades animales de declaración obligatoria El presidente de la Republica y el ministro de Agricultura y Ganadería. La Gaceta No.156. http://www.pgrweb.go.cr/scij/Busqueda/Normativa/Normas/nrm_texto_completo.aspx?n Valor1=1&nValor2=63731
Díaz, G., Rosadio, R., Marcelo, G., Chero, A., Jiménez, Ar., Reyna, I., & Maturrano, L. (2017). Evaluación de una Técnica de PCR-Múltiple para la Detección Rápida de Salmonella Typhimurium y Enteritidis en Cuyes (Cavia porcellus) Naturalmente Infectados. Revista de Investigaciones Veterinarias Del Perú, 28(3), 713–722. https://doi.org/http://dx.doi.org/10.15381/rivep.v28i3.13361
Díaz de la Osa, A., Pantoja, M. Q., & Rodríguez, A. H. (2015). Estandarización de la Reacción en Cadena de la Polimerasa con Transcripción Reversa para el diagnóstico molecular de potyvirus que afectan a Capsicum annuum L. en Cuba. Revista CENIC. Ciencias Biológicas, 46(2), 144–155.
Ebentier, D. L., Hanley, K. T., Cao, Y., Badgley, B. D., Boehm, A. B., Ervin, J. S., Goodwin, K. D., Gourmelon, M., Griffith, J.F., Holden, P. A., & Kelthy, C.A. (2013). Evaluation of the repeatability and reproducibility of a suite of qPCR-based microbial source tracking methods. Water Research, 47(18), 6839–6848. https://doi.org/10.1016/j.watres.2013.01.060
Foley, S. L., Johnson, T. J., Nayak, R., Ricke, S. C., & Danzeisen, J. (2013). Salmonella Pathogenicity and Host Adaptation in Chicken-Associated Serovars. Microbiology and Molecular Biology Reviews, 77(4), 582–607. https://doi.org/10.1128/mmbr.00015-13
García, R. (2019). Validación de una metodología para la cuantificación de un microorganismo probiótico (Lactobacillus acidophilus La3) en yogur [Tesis de maestría, Universidad de Antioquia, Colombia]. Archivo digital. http://bibliotecadigital.udea.edu.co/handle/10495/11259
Heymans, R., Vila, A., van Heerwaarden, C. A. M., Jansen, C. C. C., Castelijn, G. A. A., van der Voort, M., & Biesta-Peters, E. G. (2018). Rapid detection and differentiation of Salmonella species, Salmonella Typhimurium and Salmonella Enteritidis by multiplex quantitative PCR. Public Library of Science One, 13(10), 1–15. https://doi.org/10.1371/journal.pone.0206316
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K., von Haeseler, A., & Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nature methods, 14(6), 587-589.
Katoh, K., & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular biology and evolution, 30(4), 772-780.
Koetsier, G., & Cantor, E. (2019). A practical guide to analyzing nucleic acid concentration and purity with microvolume spectrophotometers. New England Biolabs Inc. technical note, 8, 7–19. https://www.bioke.com/blobs/downloads/NEB/MVS_Analysis_of_NA_Concentration_and_Purity.pdf
Kralik, P., & Ricchi, M. (2017). A basic guide to real time PCR in microbial diagnostics: Definitions, parameters, and everything. Frontiers in Microbiology, 8(1), 1–9. https://doi.org/10.3389/fmicb.2017.00108
Lavalett, L., Sánchez, M. M., Múñoz, N., Moreno, J., & Cardona-Castro, N. (2009). Desarrollo y validación de una reacción en cadena de la polimerasa múltiple para la identificación de los serogrupos B, C2, D y E de Salmonella entérica. Biomédica, 29(1), 244–252.
Liu, Y., Zhang, D. F., Zhou, X., Xu, L., Zhang, L., & Shi, X. (2017). Comprehensive analysis reveals two distinct evolution patterns of Salmonella flagellin gene clusters. Frontiers in Microbiology, 8(1), 1–12. https://doi.org/10.3389/fmicb.2017.02604
Lorenz, T. C. (2012). Polymerase Chain Reaction: Basic Protocol Plus Troubleshooting and Optimization Strategies. Journal of Visualized Experiments, 63(1), 1–15. https://doi.org/10.3791/3998
Minh B.Q., Nguyen, M.A.T., & von Haeseler A. (2013). Ultrafast approximation for phylogenetic bootstrap. Molecular Biology and Evolution, 30(5), 1188-1195.
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., & Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular biology and evolution, 32(1), 268-274.
Nurjanah, S., Rahayu, W. P., & Mutaqin, L. Al. (2018). Detection Method for Salmonella Typhimurium and Salmonella Enteritidis using Real-Time Polymerase Chain Reaction. International Journal of Engineering & Technology, 7(1), 302–306.
World Organization for Animal Health (OIE). (2014). Development and optimization of nucleic acid detection assays. Chapter 3.6.3. In: Manual of Diagnostic Tests and Vaccines for Terrestrial Animals. Paris, France: OIE.
World Organization for Animal Health (OIE). (2019). Principles and methods of validation of diagnostic assays for infectious diseases. Chapter 1.1.6. In: Manual of Diagnostic Tests and Vaccines for Terrestrial Animals. Paris, France: OIE.
Pegg, D. E. (2015). Cryopreservation and Freeze-Drying Protocols. Springer Protocols, 1257(1), 477–488. https://doi.org/10.1007/978-1-4939-2193-5
Rambaut, A. (2009). FigTree 2012-2014 (Versión 1.4). http://tree.bio.ed.ac.uk/software/figtree
Secretaría Ejecutiva de Planificación Sectorial Agropecuaria (SEPSA) (2020). Boletín Estadístico Agropecuario: Serie Cronológica 2016-2019. Edición Nº30. http://www.infoagro.go.cr/BEA/BEA30.pdf
Servicio Nacional de Salud Animal (SENASA). (2013). Protocolo Vigilancia de Salmonella en Aves Reproductoras. Programa Nacional Salud Aviar, versión 02. http://www.senasa.go.cr/senasa/sitio/files/231014045057.pdf
Trevethan, R. (2017). Sensitivity, Specificity, and Predictive Values: Foundations, Pliabilities, and Pitfalls in Research and Practice. Frontiers in Public Health, 5(1), 1–7. https://doi.org/10.3389/fpubh.2017.00307
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A., & Minh, B. Q. (2016). W-IQ-TREE: a fast-online phylogenetic tool for maximum likelihood analysis. Nucleic acids research, 44(1), 232-235.
Xiong, D., Song, L., Geng, S., Tao, J., An, S., Pan, Z., & Jiao, X. (2016). One-Step PCR Detection of Salmonella Pullorum/Gallinarum Using a Novel Target: The Flagellar Biosynthesis Gene flhB. Frontiers in Microbiology, 7(1), 1863. https://doi.org/10.3389/fmicb.2016.01863
Xiong, D., Song, L., Pan, Z., & Jiao, X. (2018). Identification and Discrimination of Salmonella enterica Serovar Gallinarum Biovars Pullorum and Gallinarum Based on a One-Step Multiplex PCR Assay. Frontiers in microbiology, 9(1), 1718. https://doi.org/10.3389/fmicb.2018.01718
APÉNDICE
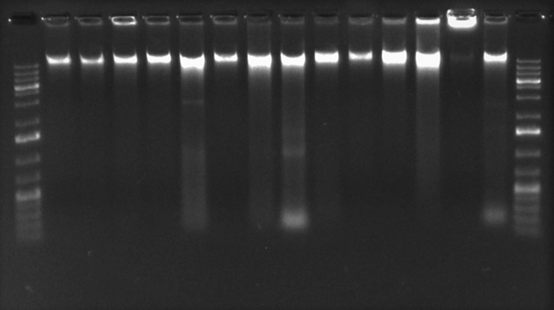
Fig. S1. Electroforesis en agarosa 1% para determinar la integridad del ADN extraído. De izquierda a derecha: marcador molecular 1kb; ADN de S. Gallinarum cepa 1, 2, 3 ,4 ,5 ,11; S. Enteritidis cepa 9, 6; S. Abaetetuba cepa 8; S. Choleraesuis cepa 7; S. Typhimurium cepa 10; P. multocida cepa 14; S. aureus cepa 12; E. coli cepa 13; control Negativo (agua doble desionizada libre de nucleasas).
TABLA S1
Datos de concentración y pureza del ADN extraído de cepas bacterianas aisladas.
|
Cepa |
Muestra aislada |
Concentración del ADN (ng/μL) |
A260/280 |
A260/230 |
|
1 |
293 (Salmonella Gallinarum) |
37,8 |
1,60 |
0,41 |
|
2 |
534 (Salmonella Gallinarum) |
21,4 |
1,51 |
0,53 |
|
3 |
535 (Salmonella Gallinarum) |
21,6 |
1,67 |
0,47 |
|
4 |
536 (Salmonella Gallinarum) |
37,6 |
1,57 |
0,59 |
|
5 |
SG 9R (Salmonella Gallinarum) |
20,0 |
1,98 |
1,46 |
|
6 |
Salmonella Enteritidis (Vacunal AVIPRO) |
14,0 |
1,66 |
0,45 |
|
7 |
Salmonella Choleraesuis (ATCC® 10708™) |
46,8 |
1,88 |
0,88 |
|
8 |
Salmonella Abaetetuta (ATCC® 35640™) |
23,6 |
1,72 |
0,62 |
|
9 |
Salmonella Enteritidis (ATCC® 13076™)) |
38,1 |
1,62 |
0,70 |
|
10 |
Salmonella Typhimurium (ATCC® 14028™) |
31,7 |
1,59 |
0,71 |
|
11 |
Salmonella Pullorum (ATCC® 13036™) |
43,2 |
2,04 |
1,72 |
|
12 |
Staphylococcus aureus (ATCC® 25923™) |
14,9 |
1,86 |
0,83 |
|
13 |
Escherichia coli (ATCC® 25922™) |
26,1 |
1,77 |
0,20 |
|
14 |
Pasteurella multocida (ATCC® 12945™) |
37,3 |
1,36 |
0,55 |
Información de las secuencias obtenidas de la base de datos Genbank, utilizadas para el análisis bioinformático de posicionamiento taxonómico de un fragmento del gen flhB (182 pb).
|
Especie |
Serotipo |
Cepa |
# Accesión GenBank |
Región extraída del Genoma (posición en pb) |
|
Enterobacter cloacae subsp. cloacae |
- |
ATCC 13047 |
NC_014121.1 |
1439040:1439426 |
|
Salmonella enterica subsp. enterica |
Enteritidis |
CP050723.1 |
2734804-2735182 |
|
|
Salmonella enterica subsp. enterica |
Gallinarum |
9184 |
CP019035.1 |
2949564-2949745 |
|
Salmonella enterica subsp. enterica |
Gallinarum |
287/91 |
AM933173.1 |
1220990-1221171 |
|
Salmonella enterica subsp. enterica |
Typhimurium |
NC_003197.2 |
2846698:2847079 |
|
|
Salmonella enterica subsp. enterica |
Stanleyville |
ARB61 |
CP017723.1 |
2030975:2031341 |
|
Salmonella enterica subsp. enterica |
Dublin |
- |
LK931502.1 |
1013219:1013584 |
|
Salmonella enterica subsp. enterica |
Pullorum |
QJ-2D-Sal |
CP022963.1 |
2871478-2871659 |
|
Salmonella enterica subsp. enterica |
Pullorum |
ATCC 9120 |
CP012347.1 |
2686037-2686218 |
Salmonella enterica subsp. enterica |
Typhi |
CMCST_CEPR_1 |
CP053702.1 |
1927865-1927980 |
Salmonella enterica subsp. enterica |
Infantis |
CVM_N17S1509 |
CP052817.1 |
2560609-2560724 |
Salmonella enterica subsp. enterica |
Anatum |
CP045518.1 |
3991839-3992217 |